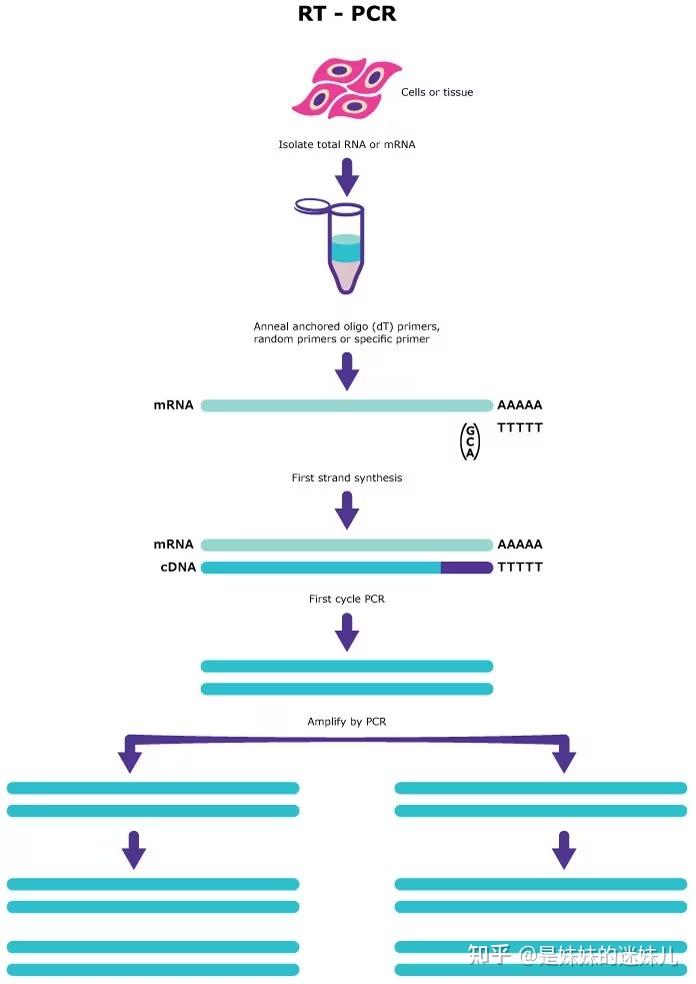
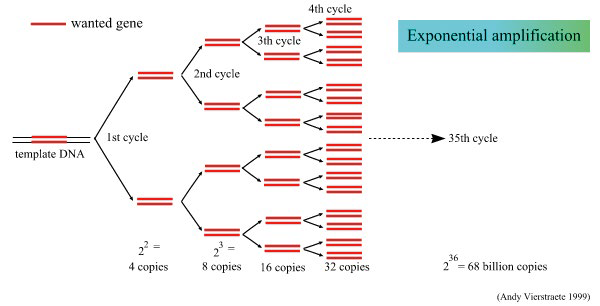
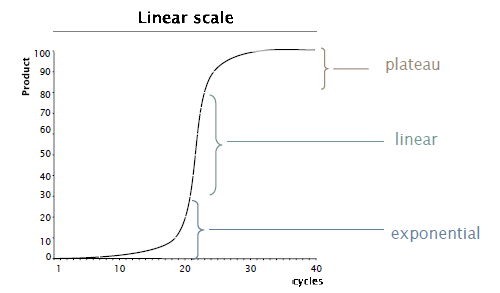
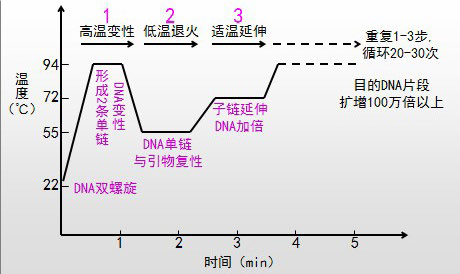
数字PCR分区方法
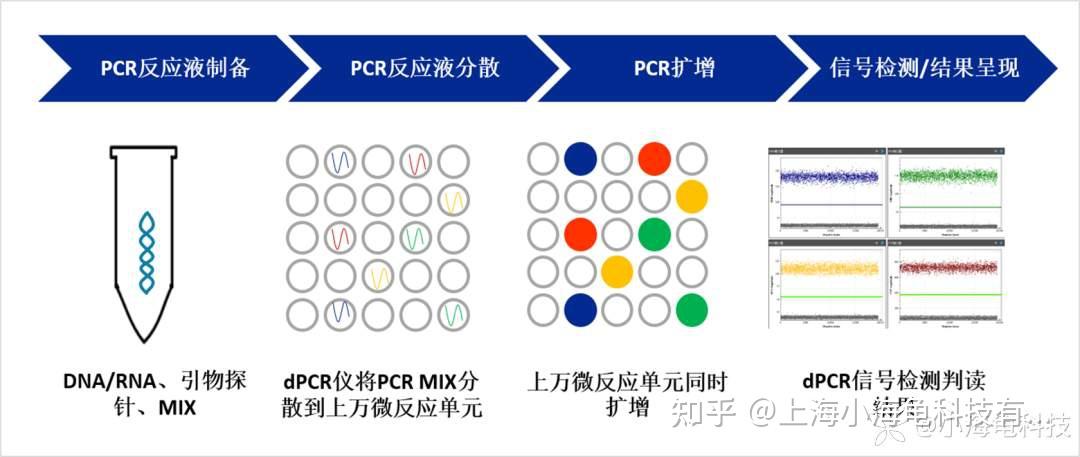
数字PCR的主要核心点在于将PCR反应液分区至数万微反应单元,实现单分子扩增。不同厂家利用不同方法实现核酸分子的分区,目前数字PCR分区方法主要包括2种:微滴数字PCR(Droplet digital PCR,ddPCR)和芯片数字PCR(Chip digital PCR,cdPCR)。分别通过微液滴和微流体芯片实现分液,分隔开的每个微小区域都可进行单独的PCR反应。
1)ddPCR技术,利用油包水微滴生成技术。2)cdPCR利用微流控芯片技术将样品的制备、反应、分离和检测等集成到一块芯片上。 数字PCR技术应用
数字PCR具有明显的优势,已经广泛应用于核酸检测的方方面面。肿瘤液态活检,遗传生殖检测,公共卫生检测,环境微生物检测,RNA表达检测,NGS文库定量,甲基化检测的等。后期将推出数字PCR具体的应用,欢迎大家持续关注。也希望大家了解数字PCR后,能够帮助大家更好地解决目前面临的困难。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-20 20:04
发表于 2024-9-20 20:04


 发表于 2024-9-20 20:05
发表于 2024-9-20 20:05