金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
美国食品药物管理局对 LDTs 的定义:
” LDTs are in vitro diagnostic products (IVDs) that are intended for clinical use and are designed, manufactured, and used within a single laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) and meets the regulatory requirements under CLIA to perform high complexity testing. ”
「LDTs 是一种临床使用的体外诊断试剂(IVDs),并且 LDTs 是由「个别」实验室「设计」、「制造」及「使用」,而此实验室需在「符合CLIA标准及规范」下执行「高度复杂的实验。」
CLIA认证是由美国医疗保险和医疗补助服务中心 (Center for medicare & Medicaid Service, CMS)颁布,并由CMS、CDC及FDA共同监管,依规定只要在美国本土使用人类检体进行临床医学检验,提供人类诊断、预防或治疗疾病或是健康评估讯息的实验室就必须符合特定联邦法规且须申请并获得CLIA的认证,而在非美国本土如有跨国合作的需求,CLIA 也提供美国海外临床医学实验室申请认证。
而依据美国食品药物管理局规定,执行 LDTs 检测的实验室必须获得CLIA’88 (Clinical Laboratory Improvement Act, 1988) 临床医学实验室认证,而获得 CLIA 认证的临床医学实验室可自行在美国全国各地进行临床检体的收样及检测,并且至少到目前为止,CLIA 认证实验室可以自行设计并执行非 IVD 认证之临床医学检验,也就是美国过去所定义的 LDTs 检验项目。
而一般在美国本土以外地區比较熟知的 CAP (College of American Pathologists,美国病理医师协会)认证,CAP 认证包含了 CLIA 认证的范围,而事实上 CAP 认证可以取代 CLIA 首次认证的的两年一次复检,两者间不同处在于 CLIA 主要重视实验室及检验结果报告的质量管理,而CAP 认证的范围除实验室本身外也包含了单项检测操作程序上的质量管理,而 因此 CAP 认证更接近于 ISO 15189 的模式。
在欧盟及韩国目前所公布的实验室认证方面,欧盟是采用 EN ISO 15189 认证;而韩国是由食品药物安全局(MFDS) 负责认证工作,但其审查的内涵仍是以 ISO 15189 为主,而台湾地区之规定与韩国相同。
回到国内的规定,先来看看国内目前对于临床医学实验室的认证,国内临床医学实验室采用多重认证方式,一般企业欲展开医学实验室业务时,需要申请「医疗机构执业许可证」,之后再依实验室执行检验项目分别申请「生物安全实验室备案凭证」、「艾滋病检测筛查实验室备案」、「临床基因扩增检验实验室技术验收合格证」、「母婴保健技术服务执业许可证」或「高通量基因测序技术临床应用试点单位资格」等,由区、市、省卫生行政主管部门负责审核许可,基本上国内对于临床医学实验室的规范标准也是以 ISO 15189 之相关规范为基础。
ISO 15189:2022 医学实验室认证标准2022年版,在国内相对应的是中国合格评定国家认可委员会 (CNAS) 的 CNAS-CL02:2023 《医学实验室质量和能力认可准则》,依目前状况来看,未来国内如实施 LDTs 管理规范,对临床医学实验室的认证与管理应该仍是依现行方法,在 ISO 15189:2022 的基础下,由省或中央级单位负责认证及许可工作,而不会委外由第三方认证单位进行认证。
但对计划在未来执行 LDTs 相关业务的公司或医学实验室来说,需要注意的是上述无论 CLIA、CAP、ISO15189 或 CNAS-CL02:2023 等认证标准,均是针对临床医学实验室,也就是采用已获得体外检测试剂许可之检验试剂进行检测的实验室,但对于 LDTs 检测的实验室而言,其使用的实验材料及相关试剂组,如探针、引物、biomarker 或标准品及缓冲液、酶等组成,多为非经体外诊断试剂许可的商业化产品,而这是该关注与及早规划的工作。
美国食品药物管理局的 The Final LDTs rule 曾多次提及生产制造的部份,而这也是影响整个 LDTs 有效性及安全性的重要关键,未来在相关法规制定的过程中,誓必会纳入相关的规划,但会采用 ISO 17025 的概念甚或是直接用GMP规范来管理,目前仍未见任何的讨论,但这部份很重要,需討論的范围也很大,接下来会再专篇讨论。
上面谈过了未来执行 LDTs 检验的实验室认证标准,所以接下来就用目前已开始施行 LDTs 管理的台湾地区来作为一个案例,探讨一下实际的执行面有那些问题。(特定醫療技術檢查檢驗醫療儀器施行或使用管理辦法:https://law.moj.gov.tw/LawClass/LawAll.aspx?pcode=L0020075 )
LDTs 检测主要的执行单位包含收集检体及作出最终诊断的「医疗机构」,以及操作检验及发出检验结果报告的「临床医学实验室」,而依据美国食品药物管理局的定义,LDTs 检测是由临床医学实验室负责设计、制造及操作,因此临床医学实验室是主要责任单位,也应是 LDTs 申请的主体,这一点亦同样可见于目前的韩国及欧盟相关规范。
但台湾地的区特管法却是以医疗机构作为 LDTs 申请的主体 (特管法第三章第二节第36条、37条),其程序为「当医疗机构有某项检验的需求,应就此检验项目的条件,寻找或建立合适的认证临床医学实验室,而后建立该 LDTs 检测的相关执行材料并提交主管机关审核」,由下表中可以看到每一项获得 LDTs 许可的检验项目后面均绑定医疗机构与认证实验室及机构名称。
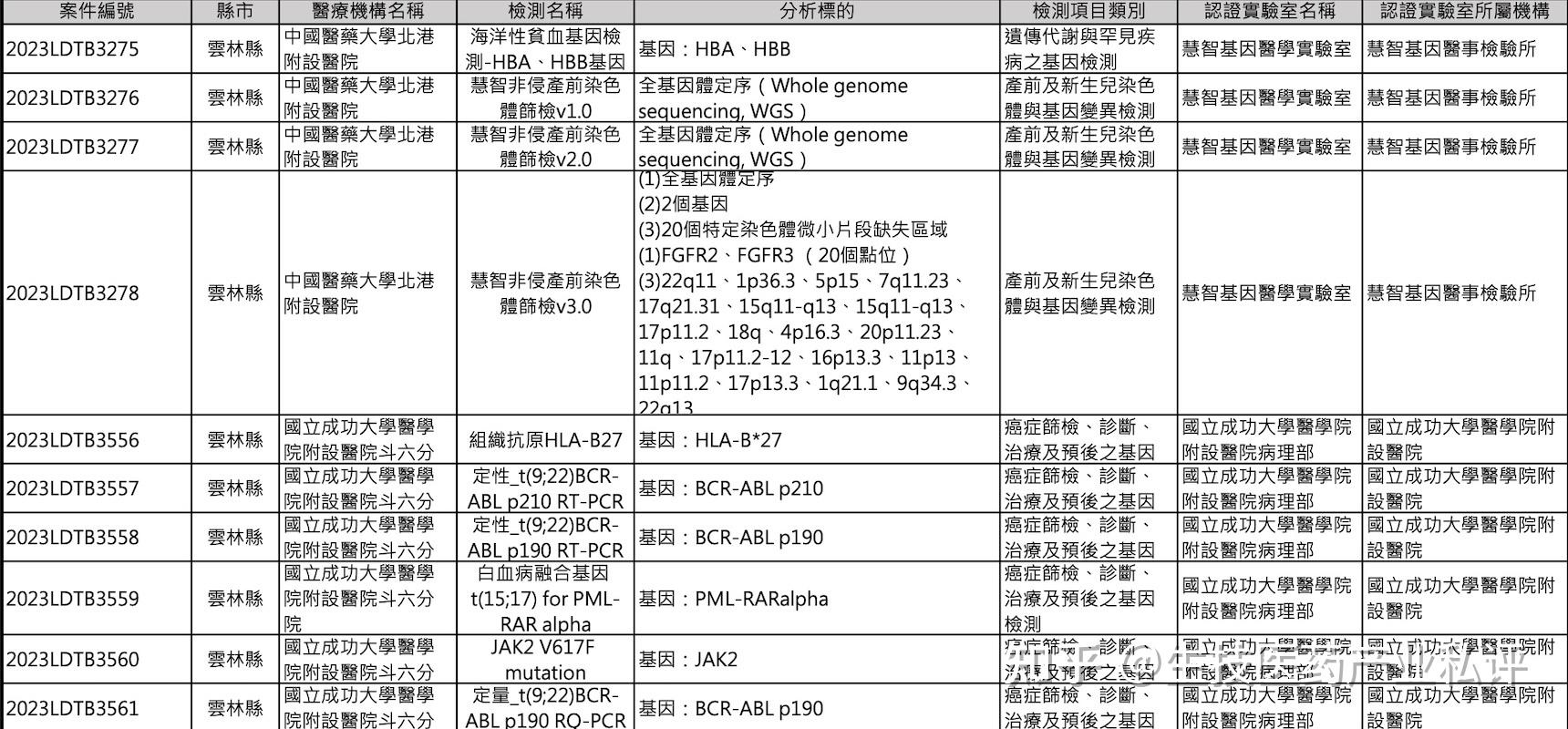
而这样设定的主要思维在于台湾地区的医师专科分类中有临床病理专科(Clinical Pathology)这一个项目 (有兴趣可以自行搜寻此专科医师的负责内容,国内前一阵子有人在推重血液病理专科的成立,也是类似的概念,在此就不多作贅述),而法规规定当检验报告具有诊断性质者,就必须有临床病理专科医师或相关专科医师的签署才能发出报告,因此也就有以医疗机构相关专科医师为主体进行 LDTs 申请的模式。
但在医学检验行业的实务操作上,除非是医疗机构院内自设的医学检验实验室 (如上表中的成功大学附设医院与附设医院病理部),一般来说,即使在台湾绝大部份的 LDTs 检测都是由商业临床医学实验室开发,然后寻求医疗机构的合作样本的收集以进行检测。
第三方检验机构是整个系统的主要操作及执行者,也是相关申报材料的准备者,且医疗机构或报告签署医师不能也不会去涉入此类商业公司的内部操作及经营管理,因此,一但检测发生问题,责任的归属问题要由签署报告的医疗单位、医生亦或是临床医学实验室来负责,其判定将造成错乱及不公。同时,采医疗机构搭配临床医学检验实验室共同申请的模式,也会有(单一检验)重覆申请,浪费相关资源的问题。
临床病理的医学专科制度在英美日等许多国家均有施行,但在欧美等地的法规规定,可签署此类报告不限于医师,还包含临床病理学家及受过一定训练的临床检验技师,因此并不会发生与台湾地区相同的状况,而国内目前并无临床病理专科的存在或有必须医师签署的相关法规,经过一定训练的临床检验技师亦可签发报告,所以未来也不会产生此类问题。
此外,由上表中可以看到每一项 LDTs 许可的检测项目均明确注明「检验名称」及「分析标的」,也就是每一项检测都需单独申请及审核,比照 IVD 的管理模式,而此已是全球对 LDTs 管理的共同趋势,目前美国、欧盟及韩国等将 LDTs 检测纳入管理的国家均采取此方法,而非过去仅认证实验室的模式,而国内未来在纳入 LDTs 管理时估计亦是采取此种模式。
接下来的篇章就要进入 LDTs 管理规范的深水区了,也就是关于 LDTs 检测的安全性与有效性的评估标准了,包含了实验材料、试剂的质量标准,实验操作的质量标准,实验操作的标准品与仪器设备的校正品等,此外对于 LDTs 有效性的临床实验评估该做到什么程度,这部份在目前各国出台的法规、规范或建议中均未见深入的讨论,但这却是对相关厂商最需要提早准备的工作,所以我们将参考各地的相关法规、认证标准及现行市场实务操作等,进行相关探讨。 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-20 18:36
发表于 2024-9-20 18:36





