金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×

(未经授权,严禁转载,违者必究)
第一章 PCR技术发展历程
第 1 节 绪论
聚合酶链式反应(Polymerasechainreaction)也就是我们通常所称的PCR反应,自1971年发明以来,已经在医学、微生物学、植物学和动物学等领域得到广泛的应用,特别是2020年新冠疫情以来,采用核酸筛查的方法寻找新冠病毒感染人群,可谓是一种快速、适用于大规模人群的筛查手段,同时被当成阳性病例确诊手段之一,自此PCR反应被广大的普通民众所了解和熟知。众所周知,PCR反应是测序、分子克隆技术的基础,PCR技术的发明将分子生物学技术向前推进了一大步。生命科学是解决人类重大生命问题的科学,PCR技术的诞生成为生命科学的生长点,使生命科学在自然科学中的位置起了革命性的变化。20世纪50年代,遗传物质脱氧核糖核酸(DNA,Deoxyribonucleicacid)双螺旋结构的发现,开创了从分子水平研究生命活动的新纪元。此后,遗传信息由DNA通过RNA(Ribonucleicacid,核糖核酸)传向蛋白质这一“中心法则”的确立以及遗传密码的破译,为基因工程的诞生提供了理论基础。随着PCR技术的发展,对PCR技术的使用从测序和分子克隆,逐渐应用于病毒、细菌、真菌和物种的鉴别和鉴定。甚至,我国的食品安全国家标准(方法标准)中也引入了PCR技术。PCR技术具有高灵敏性、快速和高通量的特点,目前在疾病诊断、致病微生物鉴定、物种鉴别等领域得到推广和应用。
第 2 节 PCR 技术发展历程
1953年4月25日,沃森(JamesDeweyWatson)和克里克(FrancisHarryComptonCrick)在《Nature》发表DNA双螺旋结构模型,自此开创了分子生物学时代,这一发现也被誉为20世纪以来生物学方面最伟大的发现,使生命科学的研究深入到分子层次。从此以后,科学家一直在探索研究DNA的新方法,新技术。1971年,印裔生物学家科拉纳(HarGobindKhorana)提出“DNA体外合成”的设想,即经过DNA变性,与合适引物杂交,利用DNA聚合酶延伸,不断重复该过程便可克隆tRNA基因。但是当时引物合成技术水平有限,尚未发现较好稳定性的DNA聚合酶,所以科拉纳的设想逐渐被遗忘,当时DNA体外扩增技术并未获得全面推广。1973年,我国台湾科学家钱嘉韵从黄石公园热泉中的嗜热细菌,海栖热孢菌(Thermusaquaticus)中分离出耐高温的TaqDNA聚合酶,并于1976年发表于《细菌学杂志》(J.Bacteriol)。
第一代传统 PCR 技术
1983年4月的一个星期五的晚上,美国化学家穆利斯(KaryBanksMullis)驾车在高速公路上飞驰,脑海中猛然闪现出DNA双链的结构,以及让DNA片段不断自我复制的想法。1984年11月,穆利斯对一49个碱基对(bp,basepair)的DNA片段进行了10个PCR循环的复制扩增,成功完成了第一次PCR实验。1985年,穆利斯阐述了PCR技术的基本原理,即在试管中模拟细胞内DNA复制,具体包括提供DNA体外合成合适的条件,即模板DNA、寡核苷酸引物、4种核苷酸(dNTP,deoxy-ribonucleosidetriphosphate)、DNA聚合酶,合适的缓冲液体系,通过DNA变性、复性及延伸的温度与时间。最初,DNA聚合酶使用的是大肠杆菌DNA聚合酶,该聚合酶不耐热,每次加热变性DNA后都要重新补加。1986年,穆利斯将钱嘉韵发现的耐高温TaqDNA聚合酶应用于PCR反应,极大地减化了PCR工作流程。
当时,PCR实验仍需纯手工操作:设置3个不同温度的水浴槽,然后按照高温变性、退火、延伸的顺序,将PCR管浸泡在不同温度水浴槽中,完成DNA变性、引物结合和DNA合成的PCR循环,完成一次PCR实验需要几十个循环,十分费时费力。1988年穆利斯所在的西特斯(PE-Centus)公司发明了第一台PCR自动化循环仪,在耐高温TaqDNA聚合酶的配合下,实现了PCR技术的实际应用。1989年,美国《Science》杂志将PCR列为十余项重大科学发明之首,并将TaqDNA聚合酶命名为“年度分子”。1993年,穆利斯因PCR技术被授予诺贝尔化学奖。
第二代定量 PCR 技术
20世纪90年代早期,罗氏(Roche)的科学家团队率先在PCR反应体系中加入溴化乙二胺,将样品置于紫外光下检测PCR终点荧光信号,可实现目标基因的定性检测。但这仍然给科学家们留下了一个问题——如何更容易地量化扩增产物的数量,从而确定他们开始扩增的DNA的数量。定量PCR(QuantitativePCR,qPCR)的概念被提出来,即在在PCR反应体系中加入荧光化学物质,随着PCR反应的进行,PCR反应产物不断累积,荧光信号强度也随之等比例增加,每经过一个循环,报告一个荧光强度信号,当报告荧光信号的数量达到一定的阈值时,可以测量相对数量的DNA(PCR产物)——越早达到阈值,样本的初始量越多。这种连续的荧光测量是当今qPCR技术的基础,又被称为实时荧光定量PCR(Real-timefluoroscencequantitativePCR,RT-PCR)。
初始的RT-PCR通过在PCR反应中使用能够与DNA结合的荧光染料,如SYBRGREEN染料,来实现对PCR过程的实时检测。SYBRGREEN与双链DNA(dsDNA)结合力远高于溴化乙二胺,结合后能够释放很强的荧光信号,提高了PCR检测灵敏度,缩短了PCR实验周期。由于SYBRGREEN释放的荧光信号强度与PCR产物数量成正比,可通过Ct值和标准曲线对样品中的DNA(或cDNA)的起始浓度进行准确定量。20多年来,以SYBRGREEN等荧光染料为基础的RT-PCR方法仍然是基础科学研究中最流行的选择之一,但仍有一些问题,包括产物特异性和扩增目标的信号的不确定性。DNA结合染料与所有双链DNA结合,无法区分非特异性引物退火或伪引物二聚体所产生的多个产品。为了弥补荧光染料的缺点,在PCR反应中,引入了第三个寡核苷酸序列,即探针(Probe),其上面标记了荧光修饰基团,与目标序列特异性结合后,可在特定波长的激发光下释放出荧光信号,信号强度与目标DNA成正比。由于探针只有与特异性序列结合后才能被激发出荧光信号,与DNA荧光染料相比,提高了荧光信号释放的特异性。
2000年日本学者Notomi在《NucleicAcidsRes.》杂志上公开了环介导等温扩增技术(Loop-mediatedisothermalamplification,LAMP)。2009年,日本荣研化学株式会社(以下简称“荣研公司”)研制出H1N1环介导等温扩增法检测试剂盒,可通过早期快速诊断对防止该病症的快速蔓延起到积极作用。目前,LAMP技术具有高特异性、高灵敏度,操作简单、对仪器设备要求低等优势,扩增结果可通过观察白色浑浊或绿色荧光的生成来判断,已被广泛应用于病毒、细菌、寄生虫等引起的疾病检测、食品化妆品安全检查及进出口快速检测。
第三代数字 PCR 技术
1999 年肿瘤基因组学专家福格尔斯泰因(Bert Vogelstein)和金兹勒(Kenneth W Kinzler) 首次在美国科学院院刊 PNAS 上提出“第三代 PCR”数字 PCR(Digital PCR, dPCR)的概念, 通过将样本分充分稀释,分配到不同的 PCR 反应单元,每个单元包含≤1 个拷贝的 DNA/RNA 模板,每个反应单元内均单独进行 PCR 扩增反应,扩增结束后对各个反应单元的荧光信号 进行统计学分析,以实现 DNA/RNA 绝对定量及稀有等位基因的检测。福格尔斯泰因和金 兹勒将该项技术应用于研究肿瘤罕见突变研究,并于 2003 年进一步提出基于小珠(Bead)、 乳浊液(Emulsion)、扩增(Amplification)、磁性(Magnetic)的 BEAMing 技术,将 dPCR 技术和流式技术进行结合,应用乳状液实现单个试管中实现 PCR 反应体系的分隔。
随着微流控芯片、油包水乳化液滴和纳米制造技术的发展, dPCR 的基本方法已经逐 渐建立。根据反应单元的不同分隔形式,形成了微反应室/孔板 dPCR(Chamber Digital PCR, cdPCR)、微流体 dPCR(Microfluidic Digital PCR,mdPCR)(大规模集成微流控芯片)和微 滴式 dPCR(Droplet DigitalPCR,ddPCR)三大体系。2006 年,Fluidigm 公司推出了第一台 商业化的基于芯片的商品化数字 PCR 系统——IFC 平台。该平台使用物理矩阵的策略,可 将 48 个样品逐个分布在 770 个微反应单元中。2013 年,Life technologies 推出 QuantStudio3D 数字 PCR 系统,采用高密度纳升流控芯片技术,将样本均匀分配至 20000 个单独的反应孔 中。BioRad 公司采用油包水乳化微滴技术,借助微滴发生器,将样品均分成数万个微液滴。 截至目前,美国 Thermo-Fisher 的 QuantStudio 3D 系统(cdPCR)和 Bio-Rad 的 QX200 系统 (ddPCR)占据了 dPCR 的绝大多数市场份额。
第二章 PCR 实验室的建设及质控
PCR 实验室建设的主要目标是为 PCR 检验提供一个安全、规范、方便、适宜的环境和场所。PCR 检 验是一项要求高、技术性较强的工作,为了确保 PCR 检验工作的顺利进行,保证 PCR 检验工作的质量, 实验室的建设,包括实验室工作区域的设置、设备配置、质量控制和质量保证、清洁及废弃物处理等非常 重要。
环境和场所作为实验室检验中非常关键的一个要素,在 PCR 检验中同样至关重要。目前,在我国相关 国家标准、行业标准、实验室认可指南、国家相关部委公告等文件中均对 PCR 实验室的设计和建设都给出 了明确的要求,把涉及基因扩增的实验室包括 PCR 实验室的建设和管理纳入了法制化、规范化的轨道。
第 1 节 PCR 实验室工作区域的设置和要求
PCR实验室的设计和布局应符合相关法律法规的要求,符合GB/T27025-2008《检测和校准实验室能力的通用要求》,为避免污染、确保生物安全必须严格遵循GB/T19495.2-2004《转基因产品检测实验室技术要求》、及GB19489-2008《实验室生物安全通用要求》、GB50346-2011《生物安全实验室建筑技术规范》等标准要求建设PCR实验室。实验室设计应将不相容活动区域进行有效隔离,合理设计实验分区,防止不同区域间的交叉污染对实验结果造成影响,并确保检测工作区域中的生物、化学、辐射、和物理危险控制在已经过评价的、适当的风险程度,应考虑意外伤害和职业病风险,并尽量将其风险降到最低,同时,还要保证所有工作人员和外来人员免受已知危害的伤害。
PCR实验室通常划分为五个工作区域:试剂贮备和准备区、样品制备区、PCR反应配制区、扩增区、扩增产物分析区。五个工作区应按照清洁、半污染到污染的顺序排列。有条件的实验室宜在各工作区域设置缓冲间,缓冲间压力应为负压(或上设抽风装置),与其相连的工作区域为正压。工作区域与缓冲间宜安装磁性连锁装置。受场所限制无条件设置缓冲间的实验室,在对检测区域进行功能划分后,应根据标准要求设置各工作区域的压力。每个区域应为独立的工作区,要设置明显的标志,如负责人的姓名、污染级别、联系人、联系电话、准入要求等,并清晰标记出国际通用的危险标识(如生物危险标识、放射性标识等)。各个区域不能直接相通,如果是紧密相连的区域,则需要安装物品传递窗。进入各个工作区域须严格遵循单一方向顺序,即只能从试剂贮存和准备区、样品制备区、PCR反应配制区、扩增区至扩增产物分析区,避免发生交叉污染。实验室应当配备相应的安全消防保障条件和措施,在使用、存放及处理放射性、爆炸性、毒害性和污染性物质时,应符合有关安全、防护、疏散、环境保护等规定。
一、试剂贮存和准备区
该工作区域如未设缓冲间,压力应为正压。本区的功能是实验室相关试剂(如核酸提取液、乙醇等) 的配置和贮存(包括商业化的试剂,如 PCR 反应缓冲液、Taq 酶和 dNTPs 等)。当试剂经过技术性验收符 合要求后,为避免经常打开保存管操作造成污染及反复的冻融,应将试剂分装贮存备用。贮存试剂的分装 体积根据实验室内一次测定所需的扩增反应数来确定。试剂应按照其贮存条件进行存放,通常为冰冻贮存。 在本区的实验操作过程中,操作者必须戴手套,并经常更换。此外, 操作中使用一次性帽子也是一个有效地 防止污染的措施。严禁用嘴吸取液体,加样器和吸头等必须经高压灭菌处理。
二、样品制备区
该区域如未设缓冲间,压力应为负压或减压,可以安装排风系统。本区的功能为待检样品的保存、核 酸的提取、贮存等。如在该区域内还需进行 RNA 检测,应辟出专门的 RNA 操作区,或安装二级生物安全 柜或外排风式的排风橱来替代。此区应远离其他实验操作区,粉碎样品时的器皿要单独使用,所有的器具 在使用前须经过彻底清洗并高压消毒,防止交叉污染,称取的待测试样品应加盖后再转移至 PCR 反应配制 区。
由于在加样操作中可能会发生气溶胶所致的污染,所以应避免在本区内不必要的走动。
用于 RNA 扩增检测样本制备好以后,应立即进行 cDNA 合成,因为 cDNA 链较 RNA 稳定,保存相
对容易。为保证逆转录反应的需要,应在样品制备区设置温育装置。
三、PCR 反应配制区
该区域如未设缓冲间,压力应为负压或减压,可以安装排风系统。本区的功能为配制、分装 PCR 主反 应混和液以及加入核酸模板。已纯化的核酸应保存于-20°C或-80°C,避免反复冻融,阳性和阴性标准物质 DNA 可调整至常用的浓度后分装并冷冻保存。对于使用巢式 PCR 进行检测的实验室,建议在此区域安装 二级生物安全柜或外排风式的排风橱,第一轮的扩增产物添加至第二轮的主反应混和液的操作可以在此区 域进行。
四、扩增区
该区域如未设缓冲间,压力应为负压或减压,可以安装排风系统。本区的功能为核酸扩增。加了 DNA 或 RNA 等模板的反应管应盖好盖子后拿到本区域。
如未设置 PCR 反应配制区,已制备的 DNA、RNA 模板或合成的 cDNA(来自样本制备区)的加入和主反应混合液(来自试剂贮存和制备区)制备成反应混合液等也可在本区内进行。在巢式 PCR 测定中,通常在第 一轮扩增后必须打开反应管,因此巢式 PCR 扩增有较高的污染危险性,第二次加样必须在本区内进行。不 能从本区再进入任何"上游"区域。为避免气溶胶所致的污染,应尽量减少在本区内的走动,如需要加样则应 在超净台(气流方向宜选择垂流式)或生物安全柜内进行。打开预处理过的反应混合液时必须防止液体溅 出,尤其是在巢式 PCR 仪扩增步骤之间。防止液体溅出的一个简单的方法是在打开反应管前快速离心数秒。
五、扩增产物分析区
该区域如未设缓冲间,压力应为负压或减压,应安装排风系统。本区的功能为扩增产物的测定。此区 是最主要的扩增产物污染来源,应远离其他实验操作区,还应注意避免通过本区的物品及工作服将扩增产 物带出。在使用 PCR-ELISA 方法检测扩增产物时,必须使用洗板机洗板,废液须收集至 1mol/L HC1 中, 且不能在实验室内倾倒,应至远离 PCR 实验室的地方弃掉。用过的移液器吸头也必须放至 1mol/L HC1 中 浸泡后再放入垃圾袋中按程序处理。
第 2 节 PCR 实验室设备配置
设备是实验室硬件建设的基础,是实验室重要技术要素之一。实验室能否准确、客观报告每一项检测 结果,正确选择使用合适的设备是前提条件。在设施和环境条件满足需要后,就要给 PCR 实验室配备检测 所需要的仪器设备。第一节讲到 PCR 实验室通常分为五个区域,每个区域都应配备专用的仪器设备,并且 应加贴明确的标识,避免不同区域内的设备,特别是微量移液器等小设备发生混淆或误用,从而造成环境 或样品污染,影响检测结果。
一、PCR 实验室常用设备种类
1. 温控设备:包括冰箱(4°C,-20°C,-80°C)、恒温水浴、恒温箱和液氮罐等。
2. 水净化设备:纯水仪,用于制作符合分子生物学使用标准的去离子水或超纯水。
3. 消毒设备:紫外灯、高压锅和干热灭菌器等。
4. 量值设备:包括各种型号的移液器、量筒和 pH 计等。
5. 离心设备:如冷冻离心机、水平离心机和掌式离心机等。
6. 电泳设备:如电泳仪和电泳槽等,用于核酸和蛋白的检测。
7. DNA 热循环仪(PCR 仪):如普通 PCR 仪、梯度 PCR 仪、荧光 PCR 仪等,用于核酸的扩增,可进 行定性和定量检测。
8. 凝胶成像系统:用于电泳结果的观察、拍照和分析。
9. 核酸蛋白分析仪:通过核酸和蛋白在紫外 260nm 和 280nm 有不同吸收峰的特性,用于核酸和蛋白的定量检测及提取的 DNA 纯度的检测。
10. 制冰机:用于制造大多数核酸和蛋白的实验操作所需的低温环境,以减少核酸酶和蛋白酶的降解。
11. 微波炉:用于一些溶液的快速加热。
12. 工作环境保障设备:生物安全柜、超净工作台等。
13. 超声破碎仪:用于组织匀浆,样品的提取。
14. 安全防护设备:用于紧急情况下实验室及工作人员的安全防护,如洗眼装置、紧急喷淋等。
二、PCR 实验室各区域设备配置
(一)试剂储存和准备区
1. 4°C和-20°C冰箱
2. 混匀器
3. 掌式离心机
4. 微量移液器(覆盖 1uL-1000uL)
5. 移动紫外灯
(二)样品制备区
1. 4°C和-20°C冰箱
2. 冷冻离心机
3. 混匀器
4. 水浴箱或加热模块
5. 生物安全柜或超净工作台
6. 超声破碎仪
7. 超声波水浴仪(如需处理大分子 DNA,应配备)
8. 纯水仪
9. 微量移液器(覆盖 1uL-1000uL)
10. 可移动紫外灯
(三)PCR 反应配制区
1. -20°C冰箱或-80°C冰箱
2. 生物安全柜或超净工作台
3. 掌式离心机
4. 制冰机
5. 微量移液器(覆盖 1uL-1000uL)
6. 移动紫外灯
(四)扩增区
1. DNA 热循环仪(PCR 仪)
2. 生物安全柜或超净工作台台(气流方向宜选择垂流式)
3. 微量移液器(覆盖 1uL-1000uL)
4. 可移动紫外灯
(五)扩增产物分析区
1. 电泳设备
2. 凝胶成像系统
3. 核酸蛋白分析仪
4. 微量移液器(覆盖 1uL-1000uL)
5. 可移动紫外灯
各个工作区域还应该配备消耗品:一次性手套、一次性吸水纸、耐高压处理的离心管和移液器吸头; 专用工作服和工作鞋、清洁用具;专用办公用品。以上这些物品应保证在各自区域内使用,不能交叉使用。
第 3 节 PCR 实验室质量控制和质量保证
一、实验前的质量控制
(一) 人员 实验操作人员应具备良好的分子生物学专业技术操作规范。PCR 都是微量操作,要想获得稳定可靠的 检测结果,操作人员就需要一定的专业技术知识与经验,要尽可能做到知其然又知其所以然。从实际工作 中来看,不同的操作者所获得的测定结果往往差异也很大,因此,人员培训相当重要,尤其是内部针对性 的培训。
(二)环境
实验室的设计和布局应符合相关法律法规的要求,生物安全方面要符合 GB19489-2008 的规定。要将 不相容活动的相邻区域进行有效隔离,合理设计实验室分区,进入各个工作区域须遵循单一方向顺序,即 只能从试剂贮存和准备区、样品制备区、PCR 反应配制区、扩增区至扩增产物分析区,防止不同区域间的 交叉污染对检测结果造成的影响。总结为一句话就是:严格遵守“各区独立、单一方向”。对影响检测质 量的区域的进入和使用,也要加以控制,非实验有关人员和物品不能随便进入。
(三) 仪器设备
仪器设备的正确使用、维护和校准是保证 PCR 结果准确的前提。对实验室中的仪器设备,如扩增仪、 离心机、移液器、生物安全柜等应建立一套规范化、标准化的操作程序。各实验区域应有专用的仪器设备, 每台设备要加贴唯一性标签,同一区域内的仪器设备、物品和工作服应有明显标记,避免与其他区域混用。
(四)试剂
PCR 实验室使用的试剂等级应为不含 DNA 和 DNase 的分析纯或生化试剂(另有规定的除外)。试剂 到达实验室后,需要经过符合性验收,关键试剂(包括核酸提取试剂、RNase、蛋白酶 K、阴性对照标准 物质、阳性对照标准物质、Taq 酶、限制性内切酶、引物、探针、菌种、阳性质粒等)应经过技术性验收, 合格后方能使用。实验用水应符合一级水的规格,去离子水的电阻需达到 18.2 欧姆。商品试剂盒应注明到 货日期,菌种、质粒、动植物细胞组织的贮存和保管应符合相关标准和法律法规的要求。所有试剂应按其 规定的贮存条件存放。实验室使用的试剂宜大体积配置、小体积分装后高压灭菌保存,不能高压灭菌的试 剂应过滤(0.22μm)除菌,PCR 主反应液、引物及探针应避免反复冻融。配置的试剂应在容器上标明试剂 名称、浓度、配置时间、保存条件、失效日期、配制人等信息。
(五)样品 实验室收到的样品时,应确认其包装完好无损;进入实验室后应加贴唯一性标识;在对样品进行混和、 样品制备和称量过程中要避免交叉污染。
二、实验过程中的质量控制
(一)防止污染
做好 PCR 检测的最重要的工作之一就是尽可能得防止污染。PCR 实验室中主要的污染来源包括:
1、样品间交叉污染:主要是由于样品在运输、储存、放置过程中处置不当,或是样品核酸在提取过 程中操作不当造成的样品间的污染。
2、试剂的污染:由于操作不当,造成核酸提取、扩增过程中各相关试剂的污染。
3、PCR 扩增产物污染:这是 PCR 实验中最主要最常见的污染,极微量的 PCR 产物污染就可造成假 阳性。最可能造成 PCR 产物污染的形式是气溶胶污染,操作时比较剧烈地摇动反应管、开盖、反复吹吸样 液都可能形成气溶胶而引起污染。
4、克隆质粒的污染:在用克隆质粒做阳性对照时,有时会出现克隆质粒的污染。
5、实验器具的污染:如移液器的污染等。
6、防止污染的方法:污染重在预防,为了避免实验室污染,获得准确、可靠的检测结果,实验室应 制定一套具体可行的污染防护措施并严格执行。防止防污的主要措施有:严格实验室分区及严格遵守实验 室工作制度,不同分区的物品不得混用;操作人员要有良好的习惯,实验操作要规范正确;实验器具和试 剂的规范使用;建立外来人员登记程序;每次实验后的清洁消毒等。
(二) 检测结果质量控制
为确保 PCR 实验室检测结果质量,在检测样品时,需要设置以下对照:
1、阴性质控对照:包括核酸提取空白对照(核酸提取过程中不加样品的空白管)、PCR 试剂对照(不 含 DNA/cDNA 模板的 PCR 扩增反应液试剂、阴性目标 DNA 对照(即为内源基因对照,是不含外源目标 核酸序列片段的模板,可以使用阴性标准物质,并与测试样品等同处理进行核酸提取及扩增)。
2、阳性质控对照:阳性目标 DNA 对照,即使用含有目标 DNA/RNA 序列片段的阳性标准物质和(或) 质粒。
三、外部质量评价测试
外部质量评价测试的主要方式包括:能力验证、测量审核、实验室间比对。凡是对外开展 PCR 检测并 出具检测报告的实验室,应按要求定期参加国家有关法定部门组织的检验项目的外部质量评价测试。
第三章 DNA(RNA)模板的提取和准备
第 1 节 核酸结构和特性
核酸是核糖核酸(RNA)和脱氧核糖核酸(DNA)的统称。
核酸是生命的最基本物质之一,是所有已知生命形式必不可少和最重要的组成物质,它广泛存在于所有动植物细胞、微生物体内,控制生物的遗 传特性,决定生物的表观性状。核酸不仅是生命个体基本的遗传物质,而且在蛋白质的生物合成上也占有 重要的位置,因而在生物的生长、遗传、变异等一系列重大生命现象中起决定性的作用。因此,研究核酸具有重要的意义。
其中,储存、复制和传递遗传信息的主要物质基础是 DNA,一些病毒如反转录病毒也 可以利用 RNA 作为遗传物质。除了作为遗传物质的 RNA 以外,其他生物体内常见的 RNA 还有 4 类,
第 1 类 RNA 在蛋白质合成过程中起着转运核糖核酸的重要作用,这类 RNA 简称 tRNA,它起着携带和转移 活化氨基酸的作用;
第 2 类 RNA 是信使核糖核酸,简称 mRNA,是蛋白质遗传信息的载体,作为模板用 于合成各种蛋白质;
第 3 类 RNA 是核糖体核糖核酸,简称 rRNA,是细胞内合成蛋白质的主要场所;
此外, 还有第 4 类 RNA,即现在已知许多其他种类的功能 RNA,如 microRNA 等。
不管 DNA 还是 RNA,作为 遗传物质的核酸都是相对保守的,因此,可以通过 PCR 等多种技术检测核酸,不仅可以了解肿瘤的发生、 病毒的感染、射线对人体的影响等,还可以实现物种鉴定、溯源、变异风险评估等。目前,核酸检测已经 成为生命科学领域最重要和最常用的方法。
1、核酸的基本结构
从分子水平上看,核酸是由许多核苷酸单体聚合成的生物大分子化合物。核苷酸分子单体是组成核酸 的基本单位,每一个核苷酸分子均由一分子含氮的碱基、一分子五碳糖(戊糖)和一分子磷酸组成。碱基 的种类有 5 种,包括腺嘌呤(A)、鸟嘌呤(G)、胸腺嘧啶(T)、胞嘧啶(C)、尿嘧啶(U)。五碳糖分为核酸是核糖核酸(RNA)和脱氧核糖核酸(DNA)的统称。核酸是生命的最基本物质之一,是所有 已知生命形式必不可少和最重要的组成物质,它广泛存在于所有动植物细胞、微生物体内,控制生物的遗 传特性,决定生物的表观性状。核酸不仅是生命个体基本的遗传物质,而且在蛋白质的生物合成上也占有 重要的位置,因而在生物的生长、遗传、变异等一系列重大生命现象中起决定性的作用。因此,研究核酸核糖和脱氧核糖,如果五碳糖是核糖,则形成的聚合物是 RNA;如果五碳糖是脱氧核糖,则形成的聚合物 是 DNA。
核酸分子大小差异很大,小的核酸分子仅有 21 个核苷酸,如小干扰 RNA,大的核酸分子含有数亿个 核苷酸,如人的染色体含有 2.47 亿个碱基对。核酸完全水解后产生嘌呤和嘧啶等碱性物质、戊糖(核糖或 脱氧核糖)和磷酸等。一分子核苷酸水解后可产生一分子核苷和一分子磷酸,每一个核苷分子又可以分解 为一分子碱基和一分子戊糖。碱基、核苷、核苷酸都是核苷酸的基本单位。在核酸的组成方面,DNA 和 RNA 有明显的区别,除了 DNA 含有脱氧核糖,而 RNA 含有核糖以外,DNA 和 RNA 中含有的碱基也有 差别,DNA 和 RNA 都含有腺嘌呤、胞嘧啶和鸟嘌呤,但 DNA 中不含有尿嘧啶,只有胸腺嘧啶,而 RNA 中不含有胸腺嘧啶,只含有尿嘧啶。在分子结构上,核酸中的戊糖和磷酸盐通过磷酸二酯键以交替链(糖 -磷酸骨架)相互连接,磷酸基团所连接的碳是糖的 3'-末端,与碳原子结合的碳是 5'-末端,这就产生了核 酸的方向性。
2、核酸的特性
核酸中因为嘌呤碱和嘧啶碱具有共轭双键,所以核酸具有紫外吸收的特性,DNA 钠盐的紫外吸收在 260nm 附近,其吸光率以 A260 表示,在 230nm 处于吸收低谷,因此可以使用紫外分光光度计对核酸进行定量及定性测定。在一定理化因素作用下,核酸双螺旋等空间结构中碱基之间的氢键断裂,变成单链的现象称为变性(denaturation)。引起核酸变性的常见理化因素有加热、酸、碱、尿素和甲酰胺等。在变性过程中,核酸的空间构象被破坏,双螺旋分子内部的碱基暴露,核酸的理化性质发生改变,导致其 A260 值会大大增加。A260 值的增加与解链程度有一定比例关系,这种关系称为增色效应(hyperchromic effect)。如果缓慢加热 DNA 溶液,并在不同温度测定其 A260 值,可得到“S”形 DNA 熔化曲线(melting curve)。 分析 DNA 熔化曲线可见,在缓慢加热前期,当 A260 值开始上升前 DNA 是双螺旋结构,随着温度的升高, 当核酸的部分碱基对开始断裂时,其 A260 数值随温度的升高而迅速增加,在上部平坦的初始部分尚有少 量碱基对使两条链还结合在一起,这种状态一直维持到临界温度,此时 A260 的数值最高,此后,DNA 分 子最后一个碱基对断开,两条互补链彻底分离,A260 数值开始下降。从 DNA 融化曲线可以看粗,DNA 变性作用是在一个相当窄的温度内完成的。因此,通常把加热变性时 DNA 溶液 A260 升高达到最大值一半 时的温度称为该 DNA 的熔解温度(melting temperature,Tm),Tm 是研究核酸变性很有用的参数。Tm 一 般在 85~95°C之间,Tm 值与 DNA 分子中 G C 含量成正比。
DNA 双链分离为 2 条单链后,在适当条件下,两条分开的单链又可以重新形成双螺旋 DNA,这一过 程称为复性(renaturation)。特别地,把热变性的 DNA 经缓慢冷却后的复性称为退火(annealing)。DNA 复性是非常复杂的过程,影响 DNA 复性速度的因素很多:如 DNA 的浓度、分子量、温度、溶液的离子强 度等。DNA 浓度高,复性快;DNA 分子大,复性慢;高温会使 DNA 变性,而温度过低可使错误配对不 能分离。最佳的复性温度为 Tm 减去 25°C,一般在 60°C左右,离子强度一般在 0.4mol/L 以上。亲代 DNA 双链分离后的两条单链均可作为新链合成的模板,在聚合酶的参与下按碱基配对原则完成 复制,复制完成后的子代 DNA 分子的核苷酸序列均与亲代 DNA 分子相同,但子代 DNA 分子的双链一条 来自亲代,另一条为新合成的链,故称为半保留复制。这种复制模式是核酸保持稳定性的主要原因。
3、核酸的提取
核酸的提取是指通过各种方法获得纯化的核酸 DNA 或 RNA 分子。正常状态下,DNA 和 RNA 两种分 子在细胞中都是以与蛋白质结合的状态存在的。不同物种的 DNA 差异较大,如真核生物的染色体 DNA 为 双链线性分子,原核生物的“染色体”、质粒及真核细胞器 DNA 为双链环状分子,部分嗜菌体 DNA 有时为 单链环状分子。RNA 分子结构也相差较大,在大多数生物体内 RNA 均是单链线性分子,不同类型的 RNA 分子可以具有不同的结构特点,如真核 mRNA 分子多数在 3’端带有 poly(A)结构。对于病毒来说,由于病 毒既有 DNA 病毒,也有 RNA 病毒,因此病毒的核酸分子可以是双链环状、单链环状、双链线状及单链线 状等多种形式。
研究核酸的前提是获得纯化的核酸,由于核酸位于细胞内或细胞核内,核酸提取首先就要破碎细胞, 使核酸释放出来,再去除与核酸结合的蛋白质、细胞浆中的多糖、脂类等生物大分子,去除盐类、有机溶 剂等杂质以及其它不需要的核酸分子,最后获得纯化核酸,以便于后期的分子生物学研究。核酸提取时应 注意:一是要尽可能使提取的 DNA(RNA)分子纯度较高,核酸纯度越高,越便于后续研究;二是在 DNA (RNA)提取过程中应尽量避免 DNA(RNA)的断裂和降解,尽量保持 DNA(RNA)分子的完整性,三 是在提取过程中要避免引入外源核酸的污染;四是在提取过程中要避免提取的核酸污染外界环境。
4、核酸提取的准备
进行核酸提取前,应准备好提取核酸的仪器、试剂和耗材,并根据待提取核酸是否来自真核生物、原 核生物、动物、植物、病毒等而采取不同的核酸提取方法。通常情况下,提取核酸的试剂往往含有刺激性 化学物质,操作过程务必做好防护措施,避免直接接触皮肤,防止吸入口鼻。进行核酸提取时应防止样品 或核酸对实验室产生污染,提取过程应在生物安全柜中进行,废液应弃于废液缸中,废液缸应套一次性塑 封袋,并倒入适量消毒剂。特别是 RNA 提取时,实验所用器具均应除去 RNA 酶,实验过程中戴一次性洁 净手套、口罩,提取的 RNA 溶液可适当加入 RNA 保护因子或 RNA 保护液,保证提取的 RNA 不会短时 间内降解。为保证核酸提取的稳定性和可靠性,实验室应建立核酸提取标准操作规程及实验室消毒规程。
第 2 节 核酸提取方法
近年来,生物学技术发展极为迅速,已发展出可以从各种生物样品中提取 DNA、RNA 或总核酸的多 种核酸提取方法,其中很多方法都被开发成了成熟的商业试剂盒, 在科学研究和医疗诊断领域广泛使用。 按照核酸提取方法的发展历程,可以将核酸提取方法可分为非磁性方法和磁性方法两大类。
1、非磁性方法
非磁性方法是指使用非磁性物质提取核酸的方法,根据提取试剂反应的介质不同,又可以分为液相法 和固相法(王淼等,2015)。液相法是指主要在液基中发生和反应的核酸提取方法,固相法是指提取核酸 时使用固相载体作为吸附核酸的方法。
a、液相法
液相法是最早使用的传统方法,根据使用液基的不同,可以将液相法分为十六烷基三乙基溴化铵 (CTAB)提取法、十二烷基磺酸钠提取法(SDS)、异硫氰酸胍-酚-氯仿提取法、氯化铯-溴化乙锭(CsCL-EB) 密度梯度离心法、煮沸裂解法和碱裂解法等(刘洪波等,2011)。
CTAB 提取法:CTAB 提取法是一种比较经典的 DNA 提取方法,主要应用于提取植物中的核酸,植 物中因含有大量多糖,因此,提取核酸时,常常将植物置于液氮中,在低温干燥状态下进行机械研磨,使 植物的细胞壁和细胞膜破碎(王锦等,1993)。CTAB 本身是一种阳离子去污剂,可与核酸形成复合物,使 核蛋白解聚,在低盐溶液中可以沉淀核酸和酸性多糖;而在高盐溶液中,CTAB 则会解离,使多糖和核酸 分离,然后使用乙醇等有机溶剂除去蛋白质、盐类等其它物质。据文献报道,在提取法中加入聚维酮(PVP), 能有效地防止多酚类物质降解 DNA(Kim CS et al.,1997),如在桑叶的核酸提取中加入 PVP 能有效的去除多糖类物质(王卓伟等,2001)。
SDS 提取法:这是在 CTAB 的基础上提出一种提取核酸的方法。SDS 为阴离子去污剂,在细胞裂解后, 加入高浓度的 SDS 可使蛋白质与核酸分离,释放出核酸,然后加入高浓度盐使蛋白质及多糖杂质沉淀,离 心去除沉淀后使用乙醇提纯核酸。研究发现在核酸提取过程中,SDS 法优于 CTAB 法,其步骤少,回收效 率高(肖冰梅等,2008)。
异硫氰酸胍-酚-氯仿提取法:该方法于 1987 年由 Chomczynski 等人首次提出,主要用于 RNA 的提取 (Chomczynski P et al.,1993)。RNA 的提取过程中最重要的是防止 RNA 被 RNase 降解,因此在实验中需 要加入高活性的 RNase 抑制剂。异硫氰酸胍是一种强蛋白质变性剂,既可以裂解细胞,也可以抑制 RNase的活性。该方法主要原理为:细胞经裂解后,加入等体积的酚-氯仿-异戊醇混合液,离心取上清水相;然 后经异丙醇沉淀,便可获得高质量的 RNA。该方法快速,便捷,产率高,能满足后续 Northern-blot 或建立 cDNA 库的要求。
碱裂解法:碱裂解法主要应用于提取质粒 DNA,其原理是质粒 DNA 和染色体 DNA 在变性时的差异 而进行分离,在碱性条件下,使用 SDS 裂解细胞,使染色体 DNA 和蛋白质变性,染色体 DNA 发生解链, 而质粒 DNA 由于是闭合环状结构,结合稳定,在 pH 调至屮性时,其便可恢复到原来状态,但染色体 DNA无法恢复,与蛋白质和其他杂质在高盐浓度下被沉淀分离,便可通过乙醇得到回收质粒 DNA(Bimboim HC et al.,1983)。该方法成本低,效果好,其纯度能满足后续实验的要求。
氯化铯-溴化乙锭(CsCl-EB)密度梯度离心法:是一种依靠离心沉降的物理分离核酸的方法, CsCl 经 过离心后能形成连续的密度梯度,因此可以用于不同密度物质的分离。EB 的作用主要是分离染色体 DNA 和质粒 DNA。其主要原理为:质粒是闭合的超螺旋结构,不易被 EB 插入,因此结合量少,密度小;而染 色体 DNA 容易被 EB 插入,密度相对较大,样品经离心后,便可分离质粒 DNA,再用有机溶剂提取去除 EB,提纯核酸(Wink M et al.,2006)。该方法分辨率高,纯化效果好,可用于大量质粒 DNA 的提取。
b、固相法
近年来,随着分子生物学以及高分子材料的快速发展,以固相载体系统为基础的核酸提取方法取得了 快速发展。相比液相核酸提取方法,固相核酸提取方法更方便,纯度、操作也更标准规范。固相核酸提取 系统一般包括细胞裂解、核酸结合、洗涤和洗脱四个步骤(Nakagawa THet al.,2006),主要通过调节溶液 中盐离子的浓度和采用离心柱离心的方式进行抽提(Tikoo S K et al.,2001)。固相载体种类很多,有二氧化硅 基质、阴离子交换树脂、层析吸附柱、玻璃颗粒等。
层析吸附柱:通过特殊硅基质吸附材料,能够特定吸附 DNA,而 RNA 和蛋白质顺利通过,然后利用 高盐低 PH 结合核酸,低盐高 PH 值洗脱,来分离纯化核酸(张丽等,2011)。
二氧化硅基质:二氧化硅基质在固相核酸提取方面应用广泛,其原理主要是依靠带正电核的硅颗粒和 带负电核的 DNA 之间的高亲和力(Karl-Heinz Esser et al.,2006)。细胞或样本经过裂解后,加入二氧化硅 悬浮液,室温静置,再经盐溶液及乙醇等洗漆后,使用 TE 缓冲液便可洗脱获取核酸。如裂解液中含有异 硫氰酸胍,便可同时提取 RNA,因为异硫氰酸胍为 RNase 强抑制剂。该方法现被用于很多商业化的试剂 盒,最常见的如血液 DNA 提取试剂盒和病毒 DNA 提取试剂盒等。
玻璃颗粒:玻璃颗粒分离核酸的原理与吸附色谱法类似。采用的也是高盐结合,低盐洗脱的方法。 Vogelstein 等人就曾通过玻璃粉从琼脂糖凝胶中提取核酸(VogelsmhnB et al.,1979),而 Marko 也使用该类 物质成功提取了质粒(Marko M A et al.,1982)。
阴离子交换树脂:就是利用了阴离子交换的原理,它主要依赖于带正电核的树脂表面 DEAE 基团与带 负电核的磷酸骨架之间的高亲和力。其实验操作一般为使用低浓度高 pH 值的盐缓冲液使核酸结合于交换 树脂上,洗涤除去蛋白质和其它杂质,然后使用高浓度低 pH 值的盐缓冲液进行洗脱分离,得到所需的核酸。交换树脂一般孔径较大,主要是为了亲和更多的基团。此方法所得核酸的纯度高,完整性好,但操作 步骤十分繁琐,很难大范围推广。
2、磁性方法
近年来,基于复合纳米技术开发的磁性微球(磁珠)越来越受到人们的青睐,该磁性微球已经广泛应 用于细胞分离、酶的固定、核酸纯化、免疫检测及疾病的诊断和治疗等多个领域(单志等,2006)。所谓 磁珠是指通过物理学、化学或生物学等方法将磁性无机粒子与有机高分子特异性结合所形成的具有一定磁 性及特殊结构的复合高分子微球(张敏等,2019)。相比于传统的核酸提取方法而言,使用磁珠提取核酸 具有传统提取方法所无法比拟的独特优势,主要有高效率、高特异性、高稳定性、自动化、简便通用、低 成本等优点(严希康等,1997),因此磁珠法在核酸检测中越来越普及。目前用于核酸提取的磁珠多种多样, 根据其表面性质的不同,分为以下几类:
1、羧基磁性微球:是指在表面覆盖有羧基活性基团的磁球。这也是核酸纯化中应用最广的一种修饰 磁球。以这种磁球为基础的核酸提取方法称为固相载体可逆化固定法(SPRI)。Hawkins 早在 1994 年就使 用这种方法成功的获得了质粒 DNA,并在 1995 年使用相同方法纯化了 PCR 产物(Hawkins et al.,1994)。虽然该方法应用广,但其作用机理较复杂,涉及到蛋白质组学等方面,目前业内流行的解释为:PEG 和 NaCl 有助于 DNA 构象的改变。PEG 能引起溶液中大分子的聚合。在 PEG 存在下,当盐浓度很低时,DNA 呈 随机卷曲状,当 PEG 浓度升高时,DNA 的构想就变成了紧凑致密状。松散卷曲状的 DNA 水溶性好,不 利于与磁球的结合,而紧凑致密状态的 DNA 刚好相反,这两个状态受 PEG 浓度的调节,很多研究表明羧 基磁球是核酸提取很好的载体,Aloha Spanov 等使用羧基磁球从各种乳制品中提取乳酸菌 DNA 并且进行 扩增检测,发现其去除 PCR 反应抑制因子的能力较强(Aloha Spanov et al.,2005)。
2、氨基磁性微球:即表面通过氨基修饰的磁球,其结合原理同二乙胺乙醇阳离子交换树脂的提取原 理类似:氨基磁球表面带正电荷,而磷酸骨架带的是负电荷,在一定盐浓度条件下,两种可以非特异性结 合,然后使用 Tis-HCL 缓冲液洗涤去除蛋白质等杂质,最后使用低浓度盐类把核酸从磁球卜洗脱下来。早在 2002 年,Brandon Yoza 就提出了该方法,他利用大量氨基活性基团的高分子聚合物,来对大肠杆菌的 基因组 DNA 进行提取(Brandon Yoza et al.,2002)。随后通过改进磁球的包裹技术制得结合效率极高的氨基 磁球,只耍 10ug 磁球,就可最大吸附 600ng 的 DNA,DNA 回收率能达到 95%;此外整个提取过程未使 用醇类物质,为醇类物质能抑制 PCR 扩增,这一点要优于羧基磁球和 Sigo2 磁球。Ota H 使用氨基磁球与 核酸动提取仪联用,从玉米中提取基因组 DNA,通过 PCR 方法检测转基因玉米(Ota H et al.,2006)。
3、Sigo2 磁性微球:其原理同传统固相硅类介质相问,简地说就是在盐低 pH 下,进行核酸吸附;而在低盐高 pH 下,进行核酸的分离(王森等,2007)。Bruce 使用磁珠对玉米中的基因组 DNA 进行了提取, 所得基因组 DNA 完整性好,纯度高。
4、其它磁性微球:除上述几种磁性微球外,还有聚乙烯亚胺改性磁球、核壳金磁球和共价偶联的复 合磁球等(王平康等,2006)。
3、新型核酸提取方法
近年来,在非磁性方法和磁性方法的基础上又发展了一些高通量或者微量提取核酸的方法,如微流控 芯片提取核酸方法,该方法是运用微流控技术(microfluidics)实现自动化核酸提取的主要平台,该技术具 有将生物、化学等实验室的基本功能微缩到一个几平方厘米芯片上的能力,可以完成样品的预处理、分离、 稀释、混合、化学反应和检测等所有步骤,因此也称为微型全分析系统。使用微流控芯片进行核酸的提取 以求的更高的提取效率和更快的提取时间也成为了目前研究人员关注的热点问题之一。
微流控芯片技术的本质是要有液体将各个单元连通,通过对液体的操作、处理来达到不同的目的(杨 杰等,2020)。微流控芯片结合多种微型器件如微泵、微阀、微储液器、微电极、微检测元件和连接器等 功能元器件,在微小尺度上实现流体的操控,构建出芯片实验室模型,从而将多种化学和生物学的过程集成到微全分析系统中。与其他分析技术相比较,微流控芯片的最大优势在于各种单元技术的灵活组合和规 模集成,实现了操作过程的自动化、检测目标的高通量和试剂的低消耗,排除人为干扰,防止污染,完成 自动高效的重复实验,还具有易于和其他技术设备集成以及兼容性好的特点不同于需要用到各种各样仪器 的常规方式,微流控芯片法所使用的材料包括高分子复合材料、石英材料、纸材料等。其中尤其是纸材料 和塑料材料,由于其成本低廉、材料来源广泛、废料易于回收等特点逐渐被研究作为核酸提取与检测的重 要材料。尤其是在某些偏远地区,无法提供足够充分的诊断条件,低成本、便携式、廉价的诊断芯片会更 容易满足在资源有限的环境中用于诊断。这些芯片通常需要与其他试剂一起,可以产生了核酸检测的良好 效果,也为目前常见的核酸提取法提供了另外的一种思路,可能有更好的应用价值。
微流控芯片的上述特征使得芯片实验由概念成为现实。这种从传统的中心实验室向芯片实验的模式转变,为许多研究者带来革命性进步,如从样品处理到检测的真正微型化、自动 化、集成化和便携化,原本需要在一个实验室完成的工作现在在一张芯片上即可完成。这不仅是生物样品 量和试剂的消耗大大降低到微升甚至纳升级,而且是分析速度几十倍甚至百倍提高,费用大大降低,从而 实现分析实验室的家庭化,以及发展便携式检测装置。
第 3 节 病毒基因组提取
病毒可以分为 DNA 和 RNA 两种,不同核酸类型应采取不同的核酸提取方法,即便同一种 DNA 病毒, 不同病毒中间基因组的大小差异较大,也应采取相应的提取方法。如乙肝病毒的基因组只有 3Kb,T4 噬菌 体的基因组为 165Kb,非洲猪瘟病毒的基因组 170-190Kb。针对含有病毒的样品提取病毒的基因组时,病 毒可能游离于细胞和组织之外,也可能存在于细胞之内,因此,提取病毒基因组提取时首先应保证病毒基 因组一级结构的完整性,减少病毒特别是 RNA 病毒容易降解的风险。因此,病毒提取基因组时,可以采 用较为温和的核酸提取方法。提取好的病毒核酸应有足够的纯度,应不存在对 DNA 聚合酶存在抑制作用 的有机溶剂及过高浓度的金属离子,同时应可能避免其他生物大分子如蛋白质、多糖和脂类分子的污染。 目前,病毒基因组的提取方法除了经典的碱裂解法以外,还可以选择 TRIzol 法、DNAzol 法、柱式法,如 果样品较多,可以选择高通量的磁珠法提取。本节分别以 RNA 病毒的新冠病毒和 DNA 病毒的非洲猪瘟病 毒为例,分别介绍了各自用磁珠法和柱式法提取核酸的流程和操作步骤。
1、磁珠法提取新冠病毒核酸(RNA)
目前用于提取新冠病毒核酸的磁珠法试剂盒试剂盒种类较多,不同生产厂家的操作步骤不同,可参照各自说明书进行操作。本节仅以济凡生物生产的核酸提取试剂(磁珠法)502-B 型为例进行介绍。
a、试剂盒组分 该试剂盒主要由 Buffer MVN、Buffer DW1P、Buffer MWP、Proteinase K、RNase Free ddH2O、FineMag Particles G 等组成。
b、样品准备 取采集的新冠病毒疑似样品或待检样品,在 56°C水浴 30 分钟,或 60-65°C水浴 20 分钟。 需要注意,病毒灭活处理时间是指标本/样品达到设定温度后的有效作用时间。每隔 10 分钟应将标本/样品 轻轻摇匀 1 次,另外,提高水浴温度可适当缩减灭活时间,但温度升高过多,可能降解病毒,从而降低敏 感性,增加假阴性的风险。
c、适用样本类型 鼻咽拭子、痰液、肺泡灌洗液、全血、血清、血浆、淋巴液、粪便标本、环境样 本、固体组织等。
d、核酸提取步骤:
(1)预先根据样本数量按照 20μl Proteinase K,10μl FineMag Particles G 和 500 μl Buffer MVN 的比例 混合,混合后加入 1.5 ml 离心管中,每管加入量为 530μl。注意:为了确保磁珠彻底重悬,请在使用前振 荡混匀。Proteinase K,磁珠和 Buffer MVN 混合后放置不应超过 1h。
(2)向离心管中加入 200μl 待测样本(样品需平衡至室温),涡旋振荡混匀 30sec,室温放置 10min。
(3)将离心管放置于磁力架上静置 30s,待磁珠完全吸附时小心去除液体。
(4)将离心管从磁力架上取下,加入 700μl Buffer DW1P,振荡混匀 30sec。
(5)将离心管放置于磁力架上静置 30s,磁珠完全吸附后,小心吸去液体。
(6)将离心管从磁力架上取下,加入 700μl Buffer MWP,振荡混匀 30sec。
(7)将离心管放置于磁力架上静置 30s,磁珠完全吸附后,小心吸去液体。
(8)将离心管保留在磁力架上,打开管盖,室温放置 5-10 min。
(9)将离心管从磁力架上取下,加入 50-100μl RNase Free ddH2O,70°C振荡混匀 2.5min。
(10)将离心管放置于磁力架上静置 1min,待磁珠完全吸附后,小心将核酸溶液转移至一个新离心管 (自备)中,并于适当条件保存。
2、柱式法提取非洲猪瘟病毒核酸(DNA)
非洲猪瘟病毒是引起猪高发病高死亡率的一种 DNA 病毒,常用的检测方法是利用荧光 PCR 方法检测非洲猪瘟病毒核酸。本节以柱式法核酸提取试剂盒(青岛立见生物科技有限公司)为例进行详细介绍。
1 样品准备 活猪采集血液、扁桃体液等液体样品,病死猪采集脾、肺、肾、淋巴结、肌肉等组织 样品,周边环境采集饲料、车辆、粪便、污水、栏舍等均可,采集样品后,液体样品可直接提取核酸,固 体样品可加入 5 倍的生理盐水研磨混匀,取上清液提取核酸。
2 离心 取处理后的样本上清 200μl,转入 1.5ml 离心管,再依次加入 20μl 蛋白酶 K 和 500μl 缓冲液 J1 混匀,室温孵育 5 分钟,即为样品裂解混合液。
3 将样品裂解混合液全部吸至吸附柱中(吸附柱放在收集管中),盖上管盖,12000rpm 离心 3min, 弃收集管内液体,将吸附柱重新放回收集管中。
4 打开吸附柱盖子,加入 700μl 缓冲液 J2,盖上管盖,12000rpm 离心 1 分钟,弃收集管内液体, 将吸附柱重新放回收集管中。
5 打开吸附柱盖子,加入 700μl 缓冲液 J3,盖上管盖,12000rpm 离心 1 分钟,弃收集管内液体, 将吸附柱重新放回收集管中。
6 12000rpm 离心 2 分钟,弃收集管,将吸附柱放入一个新的 1.5ml 离心管中。
7 打开吸附柱的盖子,向吸附膜的中间部位悬空滴加 50μl 缓冲液 J4,盖上盖子,室温静置 1 分钟, 再12000rpm 离心 1 分钟,弃吸附柱,盖离心管盖,管中液体即为待检样品核酸,可继续进行分子生物学 实验,也可保存于-20°C冰箱。
第四章 普通(常规)PCR 扩增
第 1 节 常规 PCR 技术的基本原理
常规聚合酶链反应(polymerasechainreaction,PCR)是20世纪80年代中期发展起来的体外核酸扩增技术。PCR是在体外模仿体内的DNA复制条件进行DNA大量快速复制的过程,具有特异、敏感、产率高、快速、简便、重复性好、易自动化等突出优点;可以从一根毛发、一滴血,甚至一个细胞中于数小时内将所要研究的目的基因或某一DNA片段扩增至十万乃至百万倍,获得足量的DNA供分析研究和检测鉴定。PCR的特点是每次PCR反应合成的产物都能和其模板一起作为下一次DNA合成的模板,使靶DNA片段呈指数形式增加。
最初建立PCR是为了扩增已知序列的靶基因。因为在PCR方法问世以前,要获得一个靶基因,必须建立基因文库,然后从成千上万的菌落中通过Southernblot杂交筛选含有靶基因的克隆。这样既费时又费钱,特别是在克隆真核生物基因时难度更大。自从建立了PCR方法以后,使克隆已知序列的基因变得非常容易。为了适应分子生物学的快速发展,PCR方法也得到了长足发展,现在PCR已应用到生命科学的各个领域。
一、PCR 技术的基本原理与过程
(一)PCR 技术的基本原理
PCR 是一种选择性体外扩增 DNA 片段的方法,DNA 的合成过程与细胞内 DNA 的天然复制过程类似。 通过试管反应使极少量的基因组 DNA 样品中的特定基因片段,在短短几小时内扩增上百万倍。PCR 扩增 的特异性是由与靶序列两端区域互补的寡核苷酸引物决定的,即 PCR 中 DNA 的合成过程均以两个引物特 异结合处为起点;一旦合成产生了以两个引物为端点的特异的 DNA 分子,则这种分子在以后的热循环中 理论上将以几何指数方式增加,从而最终累积的 PCR 产物绝大多数是以两个引物为端点特异的 DNA 片段。 PCR 就是体外模仿体内的 DNA 快速复制过程,只不过 DNA 的变性、引物退火以及新 DNA 链的合成均采 用温度的升降来控制,同时通过反复的加热、冷却使反应体系的温度反复进行高温-低温-中温的热循环, 使 DNA 的复制过程不断重复进行;而且由于每次新合成的 DNA 片段都和其模板一样可以作为下一次热循 环过程中 DNA 复制的模板,因此可以实现对特定核苷酸片段的指数级扩增(图 4.1)。
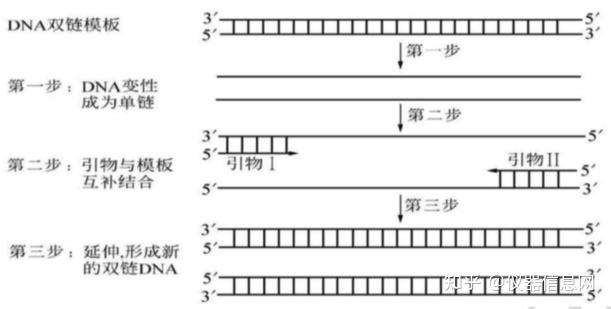
图 4.1 PCR 原理示意
(二) PCR 反应过程
PCR 进程由变性-退火-延伸三个基本反应步骤反复重复进行;1高温变性(denaturation),PCR 反应液 中的模板 DNA 加热至 93~96°C一定时间(10~30s)后,模板 DNA 双链或经 PCR 扩增形成的双链 PCR 产物 发生双螺旋的氢键断裂而变性解链变成单链 DNA,成为合成 DNA 的活性模板。2低温退火(annealing), 在反应体系温度降至特定的温度 (一般为 50~55°C)时,经加热变性成单链的模板 DNA 与引物以碱基互补 配对的方式特异性结合,形成引物模板复合物。由于引物浓度远大于模板浓度,该过程是由引物驱动的。 3中温延伸(elongation),在反应体系升温到60~75°C时单链DNA模板-引物结合物可被 DNA聚合酶识别, 并在 Taq DNA 聚合酶的作用下,以四种脱氧核糖核苷三磷酸(dNTP)为反应原料,单链 DNA 为模板,按碱 基互补配对方式,从 5'向 3'方向合成一条新的与模板 DNA 链互补的 DNA 链,使单链 DNA 模板重新成为 双链,从而完成 DNA 靶序列的半保留复制。PCR 的变性-退火-延伸三个反应步骤反复进行,使特异 DNA 片段拷贝数呈指数上升。这样经过 2~3h 可完成 30~40 个热循环,PCR 产物量最高可达起始模板量的 230 倍以上。
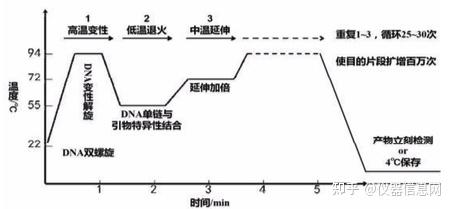
图 4.2 PCR 反应基本过程
第 2 节 PCR 扩增产物分析与处理
PCR 产物是否为特异性扩增,其结果是否符合预期,必须对其进行特定的分析与鉴定才能得出正确的 结论。目前已经发展了许多检测分析 PCR 扩增产物的方法,可依据研究对象和目的不同以及具体实验条件 而采用不同的分析方法。常见的 PCR 产物的检测分析法有凝胶电泳法、核酸探针杂交法。
一、凝胶电泳分析
凝胶电泳(gelelectrophoresis)是以凝胶为支撑物的区带电泳,常用的有琼脂糖凝胶和聚丙烯酰胺凝胶电泳等。凝胶电泳是检测PCR产物最常用、最简便的方法。通过凝胶电泳检测,我们可以判断PCR反应扩增产物的有无及大小。根据检测的目的基因不同,PCR扩增后可产生长度不同的DNA扩增片段,其核苷酸片段的分子量也有所不同,在凝胶电泳中分子量小的DNA片段移动速度快,分子量大的DNA片段移动速度慢。因此,通过凝胶电泳判断DNA扩增片段的大小,可以满足检测的需要。常用的凝胶电泳有琼脂糖凝胶电泳和聚丙烯酰胺凝胶电泳,前者主要用于DNA片段大于500bp者,后者主要用来检测小片段DNA。
(一)琼脂糖凝胶电泳的原理与方法
1.琼脂糖凝胶电泳原理
琼脂糖凝胶电泳(agarosegelelectrophoresis)是一种简便、快速、常用的鉴定、分离和纯化核酸的方法。对于分子量较大的样品,如大分子核酸、病毒等,一般可采用孔径较大的琼脂糖凝胶进行电泳分离。琼脂糖凝胶电泳的分析原理与其他支持物电泳最主要的区别是:它兼有“分子筛”和“电泳”的双重作用。
琼脂糖凝胶电泳的原理:琼脂糖凝胶电泳是以琼脂糖为介质,对不同大小的DNA或RNA实现分离的一种电泳方法。琼脂糖是一种多糖,具有亲水性,不带电荷。使得DNA在碱性条件下使其在带负电荷(缓冲液的pH8.0)在电流作用下,以琼脂糖凝胶为介质,由负极向正极移动。根据不同的DNA分子片段的大小和形状不同,在电场中泳动的速率也不相同,同时在样品中加入染料(如EB或花青素类染料)能够和DNA分子间形成络合物,经过紫外照射,可以看到DNA的位置(比对marker可知分子量大小),从而达到分离、鉴定的目的。
2.琼脂糖凝胶电泳的操作步骤
琼脂糖凝胶电泳仪的组成有,电泳仪和电泳槽两个部分,配件有与电泳槽配套使用的梳子,梳子的作用是在琼脂糖凝胶上形成点样孔,电泳仪组成见图4.3。

图 4.3 电泳仪组成
操作步骤如下:
(1)材料准备:PCR 产物、电泳缓冲液、DNA 分子量 marker (用于指示 PC 产物的分子量大小)、溴 酚蓝(用于指示电泳的进度)。
(2)制胶
琼脂糖凝胶厚度约为 3~5mm。过薄则加样孔样品会溢出来,过厚观察时荧光穿透不强以至有些小片段 DNA 带型不易分辨。如配制 2%凝胶 100ml,称取琼脂糖 2g 于三角瓶中,加入 100ml 0.5×TBE 液,微波炉 加热 2-5min,使琼脂糖完全溶解(注意不要暴沸),置室温,等温度下降至 60°C时,加入终浓度 0.5μg/ml 的 EB 液,充分混匀后倒板(注意排除气体)。
(3)加样和电泳
上样时一般取 PCR 反应液 5~10μl,加入 3μl 溴酚兰液,充分混匀,加入凝胶加样孔中。电泳仪可用一 般稳压可调中压电泳仪,电泳工作液为 0.5×TBE 液,接通电源,使样品由负极向正极移动。60-100V 恒压 电泳约 30-60 分钟。
(4)结果观察
电泳完成后,还需要经过特殊的染色才能将 DNA 条带显示出来。琼脂糖凝胶电泳结果常用的显色方法是荧光染料染色法,实验室一般采用溴化乙锭、Gold View 或 Gel Red 荧光染料染色。 溴化乙锭(Ethidium Bromide,EB)是一种荧光染料,可嵌入核酸双链的碱基对之间,在紫外线激发下,发出红色荧光,从而使肉眼无法直接观察到的 DNA 可以在紫外光照射下被观察到。琼脂糖凝胶 EB 染色 的方法可在凝胶电泳液中加入终浓度为 0.5ug/ml 的 EB,有时亦可在电泳完成后,将凝胶浸入该浓度的溶 液中染色 10~15min,于紫外透射仪上观察照相。PCR 产物经 EB 染色,紫外透射仪下观察,初步判断产物 的特异性。PCR 产物片段的大小应与预计的一致,特别是多重 PCR,应用多对引物,其产物片段都应符合 预期的大小。溴化乙锭(EB)是一种强致癌剂,长期操作 EB 或者所处实验环境中有暴露的 EB,都会对 操作人员造成潜移默化的危害,尤其是遗传方面。EB 的废液处理不当,也对周围存在生物安全隐患,污染环境。
新的核酸染料 Gel Red 可以取代 EB, Gel Red 无论用于预制凝胶染色还是凝胶电泳后染色,都表现出 了极高的灵敏度。有报道 Gold View 的毒性低于 EB,但仍然具有毒性。而 Gel Red 的特殊化学结构使其难 以穿透细胞膜进入细胞,从而降低了染料的细胞毒性,更加安全。见图 4.4。
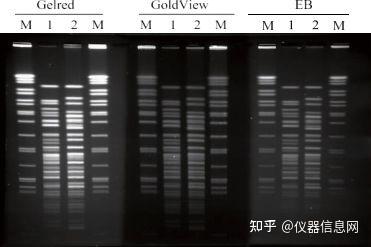
图 4.4 Gel Red、GoldView 和 EB 三种染料 PFGE 染色结果对比
(二) 聚丙烯酰胺凝胶电泳的原理与方法
1.聚丙烯酰胺凝胶电泳原理
聚丙烯酰胺凝胶电泳的原理与琼脂糖凝胶电泳类似,只不过聚丙烯酰胺 凝胶是由丙烯酰胺和甲叉双丙烯酰胺聚合而成的立体网状凝胶,其对 DNA 的有效分离范围和分辨率与琼 脂糖凝胶电泳有所不同。与琼脂糖凝胶电泳相比,聚丙烯酰胺电泳具有如下的优点:1分辨率很高,长度 仅相差 0.2%(即 500bp 中的 1bp)的 DNA 分子即可分开;2能装载的 DNA 量远远大于琼脂糖凝胶,多达 10ug 的 DNA 可以加样于聚丙烯酰胺凝胶的一个标准样品槽(1cmx1cm)而不致显著影响分辨力;3从凝胶 中回收的 DNA 纯度很高。
凝胶电泳检测电泳后的扩增产物显示的方法主要有溴化乙锭(EB)染色和银染法两种。丙烯酰胺有神经毒作用,可经皮肤吸收,并可在体内蓄积。配制和使用丙烯酰胺及亚甲双丙烯酰胺时必须戴手套和面具。 取用含上述化学药品的溶液也要戴手套。尽管可以认为聚丙烯酰胺无毒,但鉴于其中可能含有少量未聚合 的丙烯酰胺,故仍应小心处理。
2.聚丙烯酰胺凝胶电泳的操作步骤
(1)清洗
用蒸馏水将胶膜、电泳槽和梳子冲洗干净,装好胶膜,放在水平桌面上。
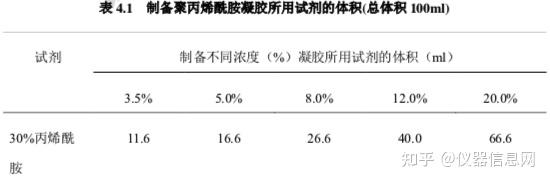
(2) 制胶
根据核酸分子质量的大小选择配置不同浓度的丙烯酰胺凝胶,一般 200-400bp 的 DNA 片段可配制 6%—8%,根据表 4.1 计算各组分的用量;在干净的三角烧瓶中分别加入上述各组分,充分混匀,小心倒 入胶膜中,插上梳子,待凝固。
(3)加样和电泳
向电泳槽中倒入 1×TBE,其量以没过胶面 2mm 为宜,小心移去梳子(如样品孔内有气泡应除去);在 DNA 样品中加入 0.2 体积的载样缓冲液,混匀后,加入样品孔内;接通电源,一般红色为正极、黑色为负 极,切记 DNA 样品由负极往正极泳动(靠近加样孔的一端为负),电压为 1-5V/cm(长度以两个电级之间 的距离计算);根据指示剂泳动的位置,判断是否终止电泳,一般 200-400bp 的 PCR 产物 100V 电压,电 泳 20-40min。
(4)结果观察
电泳结束后切断电源,取出胶膜,浸入含 0.5ug/ml 的溴化乙锭溶液中染色 10~20min;紫外透射仪上观察电泳带及其位置并与核酸分子质量标准比较被扩增产物的大小。
二、核酸探针杂交检测
(一)核酸探针杂交检测法
核酸探针杂交检测法是鉴定 PCR 扩增产物特异性的一种重要手段,常用的方法有:
1. 点杂交
点杂交是核酸杂交中比较简单的一种技术,本质上也是单链核酸分子通过碱基互补而形成 双链核酸的过程,当某单链分子被标记后,就可以检测与其互补的分子。由于杂交分子预先没有进行像 Southern 杂交和 Northern 杂交之前的电泳分离,也没有像原位杂交那样,杂交一方的分子已经表现出组织 细胞分布的位置特异性,所以点杂交的结果判断完全取决于杂交信号的强弱,其信息量没有其它杂交形式 的信息量丰富。但点杂交的操作比较简单、结果直观、适用于大批样品的检测,因此它在许多方面也得到 了广泛的应用。
2. 反向斑点杂交
利用生物素等标记的探针和特异性扩增 PCR 产物(靶 DNA 序列)杂交,但是不同 于一般的斑点杂交。一般的点印迹杂交是靶 DNA 固定于硝酸纤维素膜或尼龙膜上,标记的探针和靶 DNA 杂交显色。这种方法每次杂交反应只能判断待测 DNA 是否含有某一种探针的同源序列,对于某些基因座 位可能含有十几至几十个等位基因,用点杂交的方法就会很繁杂,甚至可能做不到。而反向斑点杂交解决 了这一难题。反向斑点杂交是先将待用的探针分别点到硝酸纤维素膜或尼龙膜上,每个探针一个点并编上 号,再将待测的 DNA 样本与之杂交,这样待测样本就会与具有同源序列的探针结合,经洗涤去除未结合 的 DNA 样本,由于待测的 DNA 样本具有生物素类的标记物,再经相应的显色反应就能显出杂交信号。这 样一次就可以判断某一基因座位生物大部分或全部等位基因。因此,可以用来鉴定扩增产物的特异性,有助 于产物的分型及法医学鉴定,且可以同时检测多个突变位点或多种病原体。
3. Southern 杂交
Southern 印迹杂交是进行基因组 DNA 特定序列定位的通用方法。一般利用琼脂糖 凝胶电泳分离经限制性内切酶消化的 DNA 片段,将胶上的 DNA 变性并在原位将单链 DNA 片段转移至尼 龙膜或其他固相支持物上,经干烤或者紫外线照射固定,再与相对应结构的标记探针进行杂交,用放射自 显影或酶反应显色,从而检测特定 DNA 分子的含量。
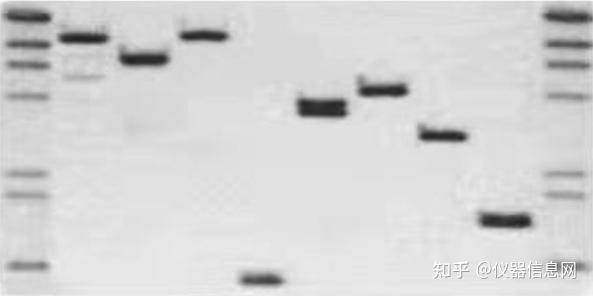
图 4.5 Southern 印迹杂交显色图片
Southern 印迹杂交技术的基本原理是:具有一定同源性的两条核酸单链在一定的条件下,可按碱基互 补的原则形成双链,此杂交过程是高度特异的。由于核酸分子的高度特异性及检测方法的灵敏性,综合凝 胶电泳和核酸内切限制酶分析的结果,便可绘制出 DNA 分子的限制图谱。但为了进一步构建出 DNA 分子 的遗传图,或进行目的基因序列的测定以满足基因克隆的特殊要求,还必须掌握 DNA 分子中基因编码区 的大小和位置。有关这类数据资料可应用 Southern 印迹杂交技术获得。Southern 印迹杂交技术包括两个主 要过程:合到一定的固相支持物(硝酸纤维素膜或尼龙膜)上,即印迹;二是固定于膜上的核酸与同位素 标记的探针在一定的温度和离子强度下退火,即分子杂交过程。
(二) 核酸探针的标记
核酸探针的标记可分为放射性标记和非放射性标记两类。
1.放射性标记
常见的放射性核素标记物有 32P、3H 和 35S。放射性核素标记的探针具有高度的特异性 和敏感性,可以检出 1~10 ug/uL 的高等生物基因组 DNA 中的单拷贝基因,然而由于放射性对环境污染限 制了其应用。
2.非放射性标记
放射性标记核酸探针在使用中的限制,促使非放射性标记核酸探针的研制迅速发展, 在许多方面已代替放射性标记,推动分子杂交技术的广泛应用。目前已形成两大类非放射标记核酸技术, 即酶促反应标记法和化学修饰标记法。
酶促反应标记探针是用缺口平移法,随机引物法或末端加尾法等把修饰的核苷酸如生物素-11-dUTP 掺 入到探针 DNA 中,制成标记探针,敏感度高于化学修饰法,但操作程序复杂,产量低,成本高。化学修 饰法是将不同标记物用化学方法连接到 DNA 分子上,方法简单、成本低,适用于大量制备(>50μg)如光 敏生物素标记核酸方法,不需昂贵的酶,只在光照 10~20min,生物素就结合在 DNA 或 RNA 分子上。
非放射性标记核酸探针方法很多,现介绍常用的几种方法如下:
(1) 生物素标记核酸探针方法:生物素标记的核苷酸是最广泛使用的一种,如生物素-11-dUTP,可用 缺口平移或末端加尾标记法。实验发现生物素可共价连接在嘧啶环的 5 位上,合成 TTP 或 UTP 的类似物。 在离体条件下,这种生物素化 dUTP 可作为大肠杆菌多聚酶 I(DNA 酶 I)的底物掺入带有缺口的 DNA 或 RNA,得到生物素标记的核酸探针。这种标记方法称为缺口平移法。用标记在 DNA 上的生物素与链霉亲 合素-酶(过氧化物酶或碱性磷酸酶)标记物进行检测。
(2) 光敏生物素标记核酸探针:光敏生物素有一个连接臂,一端连接生物素,另一端有芳基叠氮化合 物。在可见光照射下,芳基叠氮化合物可能变成活化芳基硝基苯,很易与 DNA 或 RNA 的腺嘌呤 N-7 位置 特异结合,大约每 50 个碱基结合一个生物素,所以只用于标记大于 200 个核苷酸的片段。光敏生物素的 醋酸盐很易溶于水,与核酸形成的共价结合很稳定。此法有以下优点:方法简便易行,快速省时,不需昂 贵的酶和 dUTP 等;只需光照,探针稳定,-20°C可保存 12 个月以上。适用于 DNA 和 RNA,抗体和酶等 的标记。在原位分子杂交,斑点杂交和 Southern 印迹杂交中应用,其特异性和灵敏性较高,价廉易购。
(3) 生物素-补骨脂素标记法 生物素-补骨脂素是另一种生物素光敏物质,在长波长紫外线照射下与嘧 啶碱基发生光化学反应,加成到 DNA 中,去除小分子后,得到生物素标记核酸探针。此法可标记单链或 双链 DNA 或 RNA,及寡核苷酸。灵敏度与放射性探针相当。
(4) 生物素-α-氨基乙酸-N-羟基琥珀标记化学修饰的 DNA 法 此法是在亚硫酸盐催化下,生物素酰肼 可置换寡核苷酸探针中胞嘧啶上的氨基,使生物素结合到 DNA 分子上而制成生物素化 DNA 探针。此法优 点是采用通用试剂和技术,检出敏感。
第 3 节 PCR 扩增产物纯化
PCR 反应完成目标 DNA 的扩增后,PCR 产物可以用于进一步的研究,例如用于 DNA 测序、克隆、 分析基因的功能和表达等。然而,通过 PCR 反应后反应体系中仍然存在具有活性的 DNA 聚合酶,剩余 的 dNTP、引物以及反应缓冲体系中的各种金属离子等,因此我们必须通过一定的方法从反应体系中提纯 PCR 反应的扩增产物,才有利于进一步的分析研究。
一、产物直接纯化
PCR 扩增完成后需要进行琼脂糖凝胶电泳检测,在条带单一无其他杂带的情况下,可以直接对产物进 行纯化,常见的纯化方法有:
(一)硅胶层析柱法
原理:大多数离心柱中吸附 DNA 的是一层硅胶膜,实际上就是一层玻璃纤维,其表面有大量修饰的 硅羟基(Si-OH),硅羟基在溶液中解离后带负电,然后与带正电盐离子、带负电 DNA 形成电桥,从而吸 附 DNA,使得 DNA 双链变单链,不会被生物大分子溶剂洗脱,但能经水溶性缓冲液水化后,被定量回收, 从而实现纯化分离。
(二)磁珠法
原理:磁珠是有磁性能吸附 DNA 的物质,其内部核心是铁离子,在磁场作用下可以吸附在磁体上。 在核心外,经过羧化带有羧基基团,在特定的离子条件下,可以与 DNA 结合。结合后,在外磁场的作用 下,磁珠与 DNA 结合体移动到磁体周围,可以很方便去除其他多余的液体,不能与磁珠结合的所有杂质 都随水溶液一并去除。然后,在洗脱条件下,DNA 与磁珠分离,溶解在水中,将溶解有 DNA 的溶液转移, 就得到了纯化后的 DNA 样本。
(三)其他纯化方法
1. 渗透液结合醇沉纯化
原理:利用分子大小不同,小分子的物质能够通过渗透膜溶解到大量的溶液中去,而大分子的物质不 能通过渗透膜,所以继续留存在透析袋中,通过乙醇沉淀最后达到去除小分子物质,得到纯化的有机大分 子的效果。
2. 直接沉淀纯化
原理:DNA 分子在高盐离子醇溶液中会被沉淀出来,而其他物质继续溶解在溶液中,沉淀出来的 DNA 分子经过离心与溶液成分分开,达到纯化的目的。

表 4.2
二、凝胶回收纯化
如电泳检测结果存在非特异性条带,则需要将目的条带切出来,再用胶回收试剂盒对凝胶进行回收纯 化,相较于直接纯化,只是多了一步溶胶的过程,相应的产物纯度提高,回收率则下降。
第五章 实时荧光PCR
第 1 节 实时荧光PCR原理
实时荧光 PCR(real-time PCR)是在 PCR 反应体系中加入荧光基团,利用荧光信号累积实现实时监测 整个 PCR 反应进程,对起始模板进行定量分析的方法。1995 年第一台实时荧光定量 PCR 仪由美国 Applied Biosystem 公司推出,实现了 PCR 检测由定性到定量的飞跃。与常规 PCR 相比,实时荧光定量 PCR 在反 应体系中加入荧光,提高了检测的灵敏度;引入探针,提高了反应的特异性;扩增片段多为 100-150bp, 缩短了反应时间;无需电泳检测 PCR 产物,实现了闭管检测,减少了污染发生的可能并提高了自动化程度。 此外,该技术将 PCR 方法的应用范围从核酸定性分析拓展到核酸定量分析,以及基因表达差异分析和单核 苷酸多态性(Single Nucleotide Polymorphism,SNP)分析等,成为分子生物学领域应用最为广泛的技术之 一。
1、原理:实时荧光 PCR 的原理
实时荧光 PCR 是在普通 PCR 反应体系中加入了荧光标记探针或荧光染料,随着 PCR 反应的进行,PCR 反应产物不断累积,荧光信号强度也随之增加。每经过一个循环,收集一个荧光强度信号,通过荧光强度 变化监测产物量的变化,从而得到一条荧光扩增曲线图(见图 5.1)。
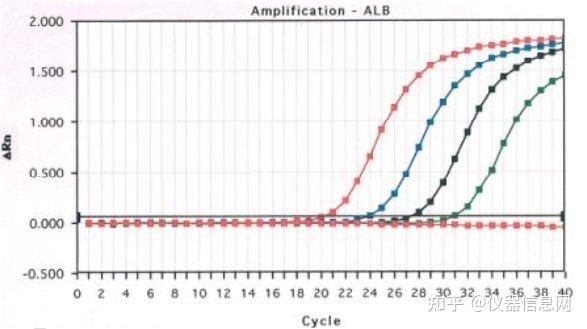
图 5.1 实时荧光 PCR 扩增曲线图。
由图 5.1 可以看出,荧光扩增曲线可大致分成三个阶段:第一个阶段是荧光背景信号阶段,在这个阶 段,产物扩增产生的荧光信号被荧光背景信号掩盖,看不出荧光信号有明显变化。第三个阶段是平台期。 在这个阶段,扩增产物达到了一定水平,已不再呈现指数级增加。用等量模板在 96 孔板上做重复实验, 收集到的荧光 PCR 扩增曲线显示,虽然起始模板量相同,但是 PCR 终产物量并不完全相同,根据最终的 PCR 产物量已不能计算出起始 DNA 拷贝数。第二个阶段是荧光信号呈指数级增加的阶段。经研究表明,
在这个阶段,PCR 产物量的对数值与起始模板量之间存在线性关系,所以选择在这个阶段进行定量分析。
2、概念:荧光定量 PCR 中几个重要概念
在介绍荧光定量 PCR 的数学原理之前,我们需要先了解几个非常重要的概念:基线、荧光阈值和 Ct 值。
基线(Baseline):是指在 PCR 的初始循环期间的信号水平,通常是 3 至 15 的循环之间,此时荧光信 号几乎没有变化。初始循环期间的低信号可以认为是背景或反应的“噪音”。
荧光阈值(threshold):是在荧光扩增曲线上人为设定的一个值,代表的是明显超出基线的扩增信号 水平(见图 5.2)。它可以设定在荧光信号指数扩增阶段任意位置上,但一般荧光域值的缺省设置是 PCR 反应前 3-15 个循环荧光信号值标准偏差的 10 倍。在阈值线上,所有样品的荧光强度与其本底荧光强度的 差值全部相同。
Ct 值:C 代表 Cycle,t 代表 threshold,Ct 值的含义是 PCR 过程中,每个反应管内扩增产物的荧光信 号达到设定的阈值时所经历的循环数(见图 5.2)。
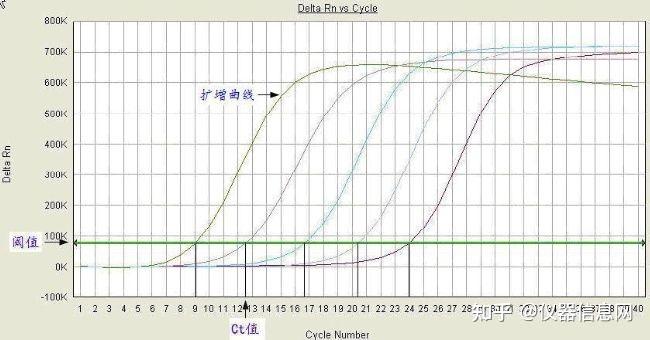
图 5.2 荧光扩增曲线中荧光阈值、Ct 值示意图。
3、数学原理:荧光定量 PCR 的数学原理:
对于普通的 PCR 反应来说,
Xn = X0 (1+E)n
其中,Xn 为 n 轮 PCR 扩增后,目的基因 PCR 产物的量;n 为 PCR 循环数;X0 为目的基因的初始模 板量,E 为 PCR 的反应效率,0≤E≤1。
由于荧光定量 PCR 反应体系中荧光物质的荧光强度与 PCR 产物的量成正比,所以用荧光强度来代替PCR 产物的量,我们可以得到:
Rn=RB+X0(1+E)nRs
式中,Rn 为 n 轮 PCR 扩增后总荧光信号强度;RB 为本底信号强度;X0(1+E)n 为 n 轮 PCR 扩增后 得到的产物量;Rs 为单位信号强度。
当循环数 n 等于 Ct 值时,所有样品荧光信号强度变化量的对数全部一致,都达到了阈值。
RT=RB+X0(1+E)CtRS
lg(RT-RB)=lgX0+Ctlg(1+E)+lgRs
Ctlg(1+E)=-lgX0+ lg(RT-RB)-lgRs
此时,如果 PCR 反应处于指数扩增阶段,所有样品的反应效率 E 稳定且近似相等;lg(RT-RB)、Rs 也 相同,那么只有 Ct 值和-lgX0 为变量,且这两个变量之间成一次性方程 y=-kx+b。也就是说,所有样品的 lgX0 与到达阈值时的循环数 n(Ct 值)呈线性关系,根据样品扩增达到阈值的循环数即 Ct 值就可计算出样 品中所含的模板量。
4、分类:荧光定量 PCR 的分类
根据所使用的荧光化学物质不同,可将实时荧光 PCR 分为使用嵌合荧光染料法和荧光探针法。
(一)荧光染料法
荧光染料法是在普通 PCR 反应体系中加入过量荧光染料。荧光染料在游离状态下基本不发光,与双链 DNA 结合后才释放出荧光信号。因此,在 PCR 体系中,随着特异性 PCR 产物的扩增,染料掺入双链 DNA 而产生的信号强度与 PCR 产物的数量是呈正相关的。
荧光染料包括饱和荧光染料和非饱和荧光染料。目前最常用的染料分子 SYBR Green I 属于非饱和荧光 染料,Eva Green 和 Solis Green 属于饱和荧光染料。两者的区别是在将 PCR 产物从 60°C逐渐升温到 95°C 制作溶解曲线的过程中,非饱和荧光染料会从已解开的单链上脱落,结合到临近的尚未解链的双链中继续 发光。饱和荧光染料不会重新与尚未解链的双链 DNA 结合,所以使用饱和染料制作的溶解曲线分辨率更高。
荧光染料法的优点是,实验设计简单,无需合成探针,降低了检测成本;操作简便,可用于监测任何 双链 DNA 序列的扩增。
荧光染料法的最大缺点在于可能会产生假阳性信号。由于荧光染料可以与任何双链 DNA 结合,无法区分不同的双链 DNA,因此非特异产物和引物二聚体的存在都会影响检测结果的准确。通过溶解曲线,虽然可以在一定程度上对双链核酸的均一性进行检测,但是精准定量分析一般还是不用荧光染料法,而使 用荧光探针法。
(二)荧光探针法
实时荧光 PCR 中使用最为广泛的荧光探针是 Taqman 探针,Taqman 技术是在 PCR 扩增体系中加入一 对引物的同时再加入一个特异性的荧光探针。该探针为一直线型的寡核苷酸,探针本身序列与目的基因两 个引物之间的某段序列互补,因此增加了反应的特异性。5’端标记一个荧光报告基团,3’端标记一个荧光 淬灭基团。探针完整时,报告基团发射的荧光信号被淬灭基团吸收,PCR 仪检测不到荧光信号。PCR 扩增 反应发生时,在退火阶段,探针与目的基因互补序列结合。在反应延伸阶段,Taq 酶的 5’-3’外切酶活性将 探针酶切降解,使报告荧光基团和淬灭基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条 DNA 链,就有一个荧光分子形成,实现了荧光信号的累积与 PCR 产物形成完全同步(见图 5.3)。Taqman 探针常用的荧光报告基团有 FAM、TET、VIC、JOE、NED 和 HEX 等,常用的淬灭基团有 TAMRA、BHQ、 MGB 和 ECLIPSE 等。
Taqman 探针根据其核苷酸序列 3’端标记的荧光淬灭基团的不同分为两种:普通的 Taqman 探针和 Taqman MGB 探针。MGB 探针的淬灭基团采用非荧光淬灭基团,本身不产生荧光,可以大大降低本地信 号的强度。同时探针上还连接有 MGB(minor groove binder)修饰基团,可以将探针的 Tm 值提高 10°C左 右。因此为了获得同样的 Tm 值,MGB 探针可以比普通 Taqman 探针设计得更短,使得探针设计的成功率 大为提高。
荧光探针法的优点是特异性高,重复性好,定量结果准确。通过合成不同荧光标记的探针,可以在一 个反应体系中实现多重 PCR 及 SNP 检测。缺点是因为需要合成探针,检测成本高。
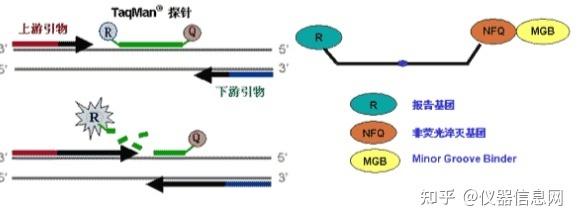
图 5.3 荧光探针法 PCR 扩增过程中发光原理示意图。
5、荧光定量 PCR 的绝对定量和相对定量
在做荧光定量 PCR 实验时,会根据实验目标确定是选择绝对定量还是相对定量。
(一)绝对定量
绝对定量是测定目标核酸分子的实际拷贝数。绝对定量实验必须使用已知拷贝数的标准品做标准曲线。 标准品可以是含有目的基因的线性化质粒 DNA,也可以是比扩增片段长的纯化后的 PCR 产物。将已知拷 贝数浓度的标准品进行梯度稀释,一般 5 个或 5 个以上梯度。将稀释的标准品和待测样品用相同的引物和 反应体系同时进行实时荧光荧光定量 PCR 扩增。标准品每个稀释度需至少三个重复,建立标准曲线。通过 待测样品的 Ct 值即可以计算出待测样品中目标核酸分子的拷贝数。
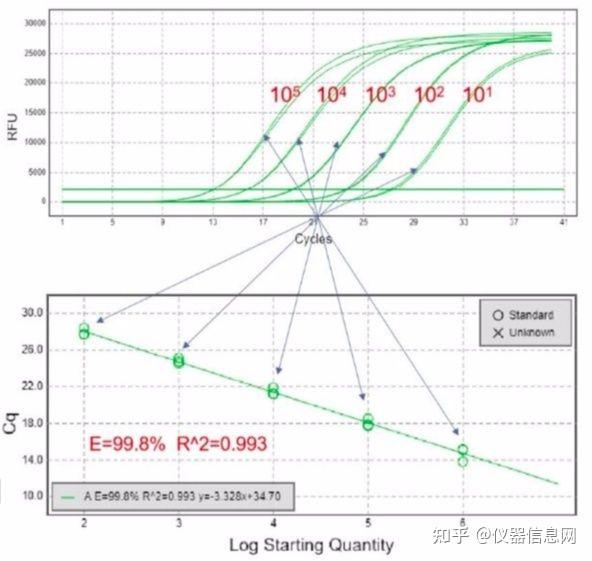
图 5.4 荧光定量 PCR 绝对定量标准曲线的建立。
(二)相对定量
在研究基因表达情况时,很多时候我们只需搞清楚该基因在不同生理阶段的变化趋势如何,无需知道 该基因的绝对量有多少。
实验操作中,由于样品选取时样品的细胞个数不可能完全相同,RNA 提取时得率不同,RNA 反转录为 cDNA 的效率不同等客观因素,用于定量分析的初始样品浓度不同。因此在进行基因表达调控研究中需 要用一些内参基因来校正因样品初始浓度不同而造成的差异。常用的内参基因是在各种生理条件下表达量 恒定的基因,这些基因也常被称为看家基因,该基因表达一般不随外界的变化而变化。常用的内参基因有 ß-Actin 基因,18S rRNA 基因等。
引入内参基因后相对定量分析的基本公式可以写成

由此派生出两种相对定量的分析方法:双标准曲线法和 ΔΔCt 法。
(1)双标准曲线法
所谓的双标准曲线法就是对每个样品的内参基因和目的基因都做绝对定量,求出每个样品中内参基因 和目的基因的绝对数量,然后根据相对定量的基本公式来求出目的基因的差异表达。
双标准曲线法的优点是考虑到了不同基因扩增效率的差异,用标准曲线来校正扩增效率,最大限度的 避免了误差;应用简便,操作灵活,无需像 ΔΔCt 法对实验进行反复的优化。缺点是每次实验都必需对目 的基因和看家基因做标准曲线。该方法适合样品量大,但是所分析目的基因较少的实验。
(2)2-ΔΔCt 法
前面我们讲过,对于普通的 PCR 反应来说, Xn = X0 (1+E)n
当循环数n=Ct,扩增效率 =1时,
添加图片注释,不超过 140 字(可选)
2-ΔΔCt 法的具体计算步骤是:首先用每一组待测样本目的基因平均 Ct 值减去待测样本看家基因平均 Ct 值,得到△Ct(待测样本);然后,用对照组目的基因平均 Ct 值减去对照组看家基因平均 Ct 值,得到△Ct (对照组);用待测样本的 △Ct 减去对照组样本的△Ct,得到△△Ct;对得到的结果取相反数,得到-△△Ct, 最后计算 2-ΔΔCt 就可以得出待测样本相对对照组的目的基因表达变化。
该方法的前提条件是目的基因和内参基因的扩增效率相同且都为 1,在实验开始前必须分别对目的基因和内参基因作标准曲线,看两者扩增效率的差别,假如两者扩增效率之间的差异小于 0.1,就可以用该方法分析。而且在接下来的实验中,无需再做标准曲线。
第 2 节 实时荧光 PCR 引物和探针设计
在第一节我们已经介绍过,实时荧光 PCR 可以分为荧光染料法和荧光探针法。荧光染料法只是在普通 PCR 反应体系中加入了荧光染料,不涉及探针,其引物设计和普通 PCR 引物设计原理是相同的。在此, 我们主要介绍使用荧光探针法时,Taqman 探针和引物的设计原理。
设计引物和探针时,应先选择好探针,然后设计引物。首先需要寻找目的基因片段,确定种内高保守、 种间高特异的区域作为引物探针设计的靶序列,尽量选择具有最小二级结构的扩增片段,因为二级结构会 影响反应效率。扩增片段长度最好在 50 bp – 150 bp,GC 含量为 30 - 80%,扩增片段越短,扩增效率越高。 无论是引物还是探针,应避免重复的核苷酸序列,尤其避免 4 个以上连续的 G;引物探针设计好后应互相 进行配对检测,以避免二聚体和发卡结构的形成。
具体来说,探针设计应尽可能遵循以下原则:(1)探针位置应尽可能地靠近上游引物;(2)普通 Taqman 探针长度应在 15-45bp(最好是 20-35bp),MGB 探针长度应在 13-25bp 以保证特异性(;3)Tm 值在 65-70°C, 通常比引物 Tm 值高 5-10°C,GC 含量在 40%-70%之间;(4)探针的 5’端第一个碱基应避免使用鸟嘌呤 G, 因为 5'G 会有淬灭作用。如果选择 FAM 标记,5’ 端的第二个碱基也不能是 G;(5)探针中,碱基 C 的 含量要明显高于 G 的含量;(6)避免探针序列中连续出现 6 个 A;(7)避免 3’端的前 4 个碱基里含有 3 个或以上的 G;(8)避免探针的中间区域含有两个或以上的 C;(9)设计好的探针应做 BLAST 分析, 保证其特异性。
引物设计应考虑:(1)引物序列应在基因的保守区段;(2)上游引物应尽可能靠近探针;(3)避 免引物自身或与引物之间形成 4 个或 4 个以上连续配对,避免引物自身形成环状发卡结构;(4)典型的 引物长度为 18-24 bp,Tm 值在 55-65°C,GC 含量在 40%-60%;(5)引物之间的 Tm 相差避免超过 2°C; (6)引物的 3’端避免使用碱基 A,避免出现 3 个及以上相同的碱基;(7)引物末端(最后 5 个核苷酸) 最好不要超过 2 个 G 和 C。
引物探针设计原则虽然很多,但是实际上大部分的引物探针设计都是专业软件完成的。比较常用的软 件有 PrimerExpress 和 Primer Premier5。我们需要做的是确定靶基因序列并导入软件,设置相应的参数就可 以了。在实际操作中可以同时设计几对引物和探针,通过实验验证引物和探针的实际使用效果。
第 3 节 不同品牌荧光 PCR 仪之间的区别
1992 年,日本人 Higuchi 首次发现实时荧光定量 PCR 技术。他在原有的普通 PCR 仪的基础上,增加 了一个激发和检测装置,并通过在 PCR 反应液中添加染料溴化乙锭,实时监测整个 PCR 反应的全过程。 1995 年美国 Applied Biosystems 公司推出了首台 PCR 仪。经过多年的发展,荧光 PCR 仪已从最初的只能 检测单一波长光源发展到可以检测多种波长光源,检测通量也从 48 个增加到 384 个。虽然目前我国也有 国产化的荧光 PCR 仪,但是市场占有率最高的定量 PCR 仪仍是进口产品,厂商依次是 ABI、罗氏(Roche) 和伯乐(Bio-rad),因此我们主要介绍这三个厂家的仪器。
在选择荧光定量 PCR 仪时,我们需要考虑仪器使用的广泛程度、仪器的检测通量、仪器的荧光通道数量、耗材的开放性、软件设计是否友好、运行速度和使用的灵活性等因素。
1、美国ABI
1995 年,ABI 公司推出了第一台定量 PCR 仪,型号为 7700。 2000 年推出第二代产品 7900 型,2004 年推出第三代荧光定量 PCR 仪 7300 和 7500 型,2007 年推出第四 代荧光 PCR 仪 StepOne 和 StepOne Plus。
7700 作为第一代产品,已经被市场淘汰,因此不再介绍。7900 是 ABI 的第二代产品,它兼容 96 孔板、 384 孔板和 Taqman 低密度表达谱芯片。可以选用手工进样或通过自动加样装置连续进样,且自动装置可 容纳 84 块 384 孔板,机械手臂可按一定顺序将待测样品板运送至主机内,配备的条形码阅读器可自动识 别进入主机的样品板编号,支持 24 小时无人操作。核心应用包括使用基因表达定量和单核苷酸多态性分 析,配套相应的分析软件。热循环系统是基于珀尔帖效应的循环系统,具有样品加热块互换功能,这一功 能增加了仪器使用的灵活性。光学检测系统为 488 nm 氩离子激光激发光源,激发光通过双轴同步扫描头 分布到所有反应孔,通过光栅和冷 CCD 照相机检测荧光。由于采用更为先进的荧光检测技术,7900 在不 降低速度、分辨率的情况下有效提高了产率。一束激光扫描并激活最多 384 个孔的每一个孔里的荧光染料, 荧光信号通过光栅分光,经过分离的多色荧光同时到达 CCD 摄像机,实现多色荧光同时检测。仪器安装 时已校准了 FAM、VIC 和 ROX 三种染料。还可兼容SYBRGreen、JOE、NED、TAMRA 和 TET 染料。半 导体加热模块,加热块 96 孔快速反应板模式下升温速率为 4.8 °C/秒,其他反应板模式下速率为 2 °C/秒。 标准模式下样品的升温速率为 1.6 °C/秒,快速模式下升温速度为 3 °C/秒。反应运行时间最短可压缩至 40 分钟。仪器检测灵敏度高,能够在 99.7 %的置信度下区分 5000 和 10000 模板拷贝数。该仪器的缺点是价 格昂贵,性能比不上 7500,占用空间大,仪器尺寸为 72 cm ×84 cm ×64 cm。
7500 是 ABI 的第三代产品,支持 96 孔板、单管和 8 连管,不支持 384 孔板。热循环系统是基于珀尔 帖效应的循环系统。96 孔加热块极限模式下升温速率为 5.5 °C/秒。标准模式下样品的升温速率为 1.6 °C/ 秒,快速模式下升温速度为 3.5 °C/秒。采用卤钨灯作为光源,卤钨灯光通过五色光源滤光片后激发每个分 析样品。卤钨灯光源增加光源滤光片后改善了长波长红色荧光标记的激发效果,提高了红色荧光检测的灵 敏度和准确性。五色荧光滤光片能够有效地分辩包括:FAM / SYBRGreen、VIC / JOE、NED / TAMRA / Cy3、 ROX / Texas Red / Cy5 在内的多种荧光染料。上述染料在安装时已经过校准。最多可支持四重 PCR(ROX 占用一个荧光通道)。仪器检测灵敏度高,能够在 99.7 %的置信度下区分 5000 和 10000 模板拷贝数。20 μL 反应体积单一报告荧光 Taqman 实验中可检测低至 10 个起始拷贝数的模板,置信度达 99.7 %。该仪器尺寸 为 34 cm ×45 cm ×49 cm,明显较 7900 小。
StepOne Plus 是 ABI 推出的第四代荧光 PCR 仪,体积很小,仅为 24.6 cm ×51.2 cm ×42.7 cm,价格较 低,适用于各种实验室环境。支持 96 孔板,热循环系统采用珀尔帖效应系统,加热块为 Veriflex 加热块, 具有 6 个独立控温区,可同时扩增 6 个不同退火温度的 PCR 产物。加热块最高升温速率为 4.6 °C/秒,样 本快速升温速率为 2.2 °C/秒。光学系统采用 LED 激发光源,4 色荧光检测系统,安装时已校正的染料有 FAM、VIC、JOE、NED、TAMRA、SYBRGreen 和 ROX。支持单机运行模式,LCD 触摸屏控制,支持 USB 存储,可实现无 PC 操作。可以进行标准模式和快速模式 PCR 反应,快速模式可将反应时间缩短至 40 分钟。
2、罗氏诊断
罗氏公司的荧光 PCR 仪型号为 LightCycler,目前已上市的型号有 LightCycler 1.5、LightCycler 2.0、 LightCycler 480、LightCycler 96 和 LightCycler Nano 等。与 ABI 仪器不同,LightCycler 采用 Therma-Base 热循环技术,热传导速率可达 20°C/秒,升降温速度快,大大缩短了仪器运行时间。反应时间短是罗氏荧 光 PCR 仪最大的优点之一。使用 LED 冷光源,维护成本降低,寿命长达 2 万小时以上。改良光路设计, 确保检测的均一性,彻底消除了边缘效应。不需要使用 ROX 校正,从而相较 ABI 荧光 PCR 仪,在荧光通 道相同的时候可以多一种荧光标记。
Lightcycler 2.0 检测样品量为 32 个,荧光检测通道由 Lightcycler 1.5 的 3 通道增加到 6 通道,提供 530 nm、560 nm、610 nm、670 nm 和 710 nm 的荧光检测。Lightcycler 96 是一款针对中通量实验需求开发的仪 器,检测样品量为 96 个。Lightcycler 480 是针对高通量实验需要而设计开发的创新一代 96 / 384 互换式荧 光 PCR 系统,秉承了 Lightcycler 系列一贯的高速、准确的特质,采用革新的 Therma - Base 模块加热技术 和独特快速散热装置,确保了孔间温度的均一性;同时匹配以先进光学检测系统,确保信号的整体采集和 检测灵敏度最大化,提供 5 色激发通道和 6 色荧光检测通道并具有颜色补偿功能。
下表是在网上查到的常见几种荧光 PCR 仪,如 Roche LightCycler96、ABI 7500fast、Bio-rad CFX96、 Stratagene Mx3000P 、Qiagen Rotor-Gene Q 的性能对比,供参考。

表 5.1 Roche LightCycler96、ABI 7500fast、Bio-rad CFX96、Stratagene Mx3000P 、 Qiagen Rotor-Gene Q 五种荧光 PCR 仪性能对比

表 5.1 续 Roche LightCycler96、ABI 7500fast、Bio-rad CFX96、Stratagene Mx3000P 、 Qiagen Rotor-Gene Q 五种荧光 PCR 仪性能对比
第六章 环介导等温扩增(LAMP)
第 1 节 技术原理
环介导等温扩增(loop-mediated isothermal amplification,LAMP)是 Notomi 等人于 2000 年提出来的一种新的核酸扩增技术,已被广泛地应用于病原微生物检测和传染性疾病诊断等 领域,显示出了令人鼓舞的应用前景。
LAMP 技术利用一种具有高度链置换活性的 Bst DNA 聚合酶,较传统核酸扩增技术具 有以下优势:
(1)等温条件下扩增,合理避免了常规 PCR 对温度循环的特殊要求所带来的 种种不便。
(2)高效灵敏,在 45~60 min 的时间内,扩增效率可达到 109~1010 个数量级, 扩增模板极限仅为几个拷贝。
(3)特异性强,采用 4 条引物,识别靶基因的 6 个位点,保证 扩增的特异性。
(4)费用低,不需要昂贵的精密仪器和特殊试剂。
(5)操作简便,不需要进 行双链 DNA 的预变性,在一管内即可以完成全部检测。
(6)检测简单,在核酸大量合成时, 产生副产物—焦磷酸镁沉淀,有极高的特异性。只要用肉眼观察或浊度仪检测沉淀浊度就能 够判断扩增与否。
(7)扩增 RNA 模板,反应体系中加入逆转录酶,即可一步实现 RNA 的 高效扩增。LAMP 反应可分为复制起始、循环扩增,延伸三个阶段,具体过程如下:
首先,是复制起始阶段。由于双链 DNA 在 65°C条件下处于动态平衡稳定状态,一条 LAMP 引物可以跟双链靶基因中的互补序列结合,继而利用 DNA 聚合酶的链置换活性启动 DNA 合成,从而取代并释放另一条单链 DNA。因此,LAMP 不同于 PCR,它不需要通过热 变性来促进双链 DNA 转化为单链。如图 6.1 中所示,上游内部引物 FIP(Forward Inner Primer) 与释放的单链 DNA 模板(1)结合,启动 DNA 的合成。在 DNA 聚合酶链置换活性作用下, 自 FIP 的 3’端 F2 处开始,合成了一条与模板 DNA 序列互补的 DNA 链(2)。
上游外部引物 F3(Forward Outer Primer)比 FIP 短几个碱基,且浓度比 FIP 低,它缓 慢地与目标 DNA 中的 F3c 互补配对(3),启动链置换 DNA 合成,最终再次形成双链模板 DNA(4)。释放 FIP 连接的互补链(5),其 5’端 F1c 与 F1 区域互补,形成颈环结构。
FIP 连接的互补链(5)作为模板供下游内部引物 BIP(Backward Inner Primer)结合并 启动 DNA 合成。继而下游外部引物 B3(Backward Outer Primer)与 B3c 区互补结合,在链 置换 DNA 聚合酶的作用下引导双链 DNA 的合成(7),并释放 BIP 引导合成的哑铃状互补 链(8),该链即为 LAMP 反应进入循环扩增阶段的起始材料。
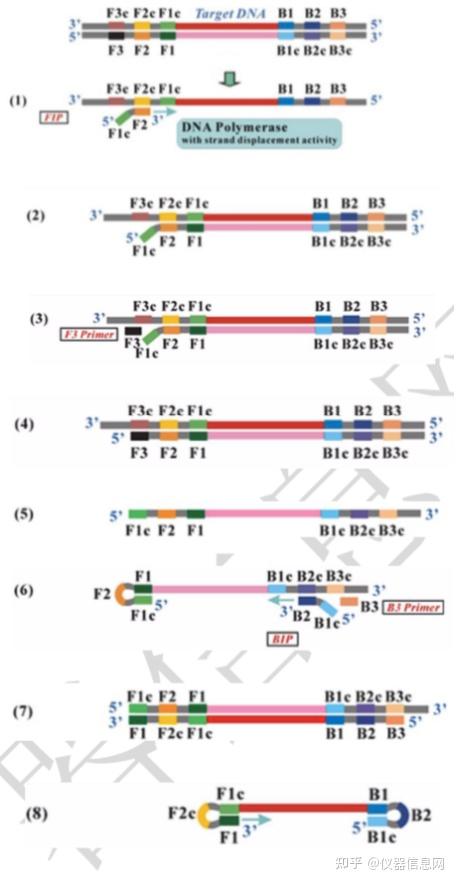
图 6.1 LAMP 复制起始阶段示意图
其次,是循环扩增阶段。如图 6.2 中所示,哑铃状 DNA 链(8)通过 F1 区 3’端自身 引导的 DNA 合成迅速转化为颈环结构。FIP 与茎环状 DNA 的单链区 F2c 结合,开始链置换 合成,解离出之前的合成链,这条释放的单链 DNA 由于 3’端存在 B1c 和 B1 互补区,从 而也会形成颈环状结构(9),继而以该链 3’端 B1 区作为起始位点,继续引导以自身为模 板的 DNA 合成(9),并释放之前 FIP 引导合成的互补链。此次释放的单链 DNA 由于两端 分别存在 F1-F1c 和 B1-B1c 两个互补区,因此形成哑铃状结构(11),恰好是(8)的翻转结 构。类似于从(8)到(11)的过程,哑铃状结构(11)迅速以 3’末端的 B1 区段为起点, 以自身为模板合成 DNA。进而 BIP 与 B2c 区结合,引导链置换 DNA 合成,释放之前由 B1 引导合成的 DNA 链。随后产生与(9)和(10)相似的结构,以及哑铃状 DNA(8)。由此 形成循环扩增。
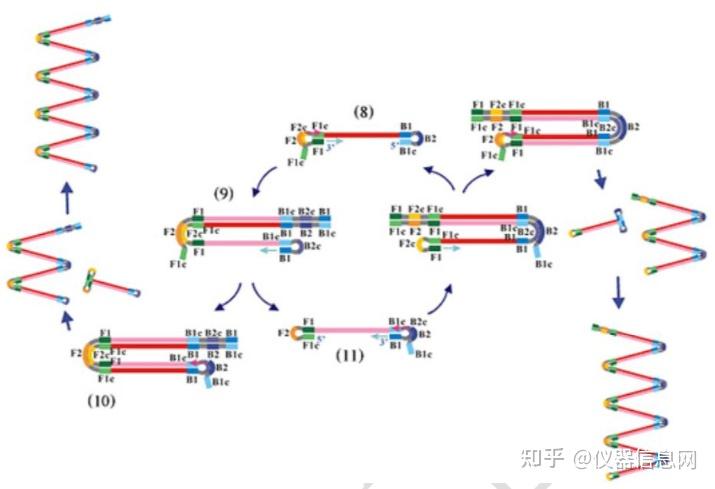
图 6.2 LAMP 循环扩增阶段示意图
最后,延伸阶段。如图 6.3 所示,由颈环状 DNA(10)为模板,BIP 与单链 B2c 区结 合,启动链置换 DNA 合成,形成颈环 DNA(12),继而以其 3’端 B1 区为起始位点,引导 以自身为模板的链置换 DNA 合成,产生长短不一的 2 条新茎环状结构的 DNA(13)和(16)。 BIP 引物上的 B2 与颈环 DNA(13)杂交,启动新一轮扩增,且产物 DNA 长度增加一倍。 因此,扩增的最后产物是具有不同个数茎环结构、多环花椰菜样结构的 DNA 的混合物。且 产物 DNA 为扩增靶序列的交替反向重复序列。
在反应体系中还可以添加环状引物 LF(Loop Primer Forward)和 LB(Loop Primer Backward),它们可以跟哑铃状结构 5’端的环状区(位于 B1 和 B2 之间,或者 F1 和 F2 之 间)进行结合,从而增加 DNA 合成的起始位点。如图 6.4 所示,其中含有 6 个环状结构的 扩增产物,在基本的 LAMP 方法中,有 4 个环是无法利用的,但加入环引物后,所有的单 链环都可以作为 DNA 合成的起始位点。因此,环引物可明显增加 LAMP 扩增的效率,缩短 反应时间。大量研究结果显示环引物可将扩增时间缩短 1/3 至 1/2,30min 内即可出现阳性 扩增(图 6.5)。
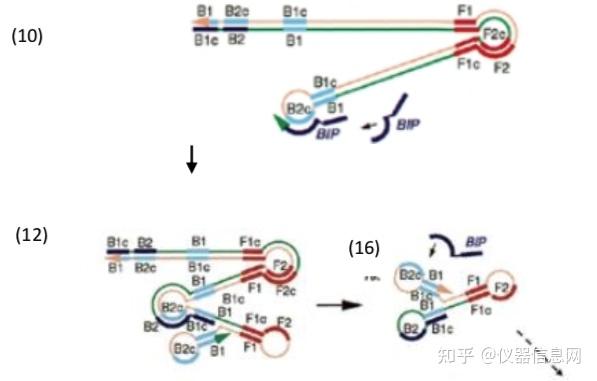
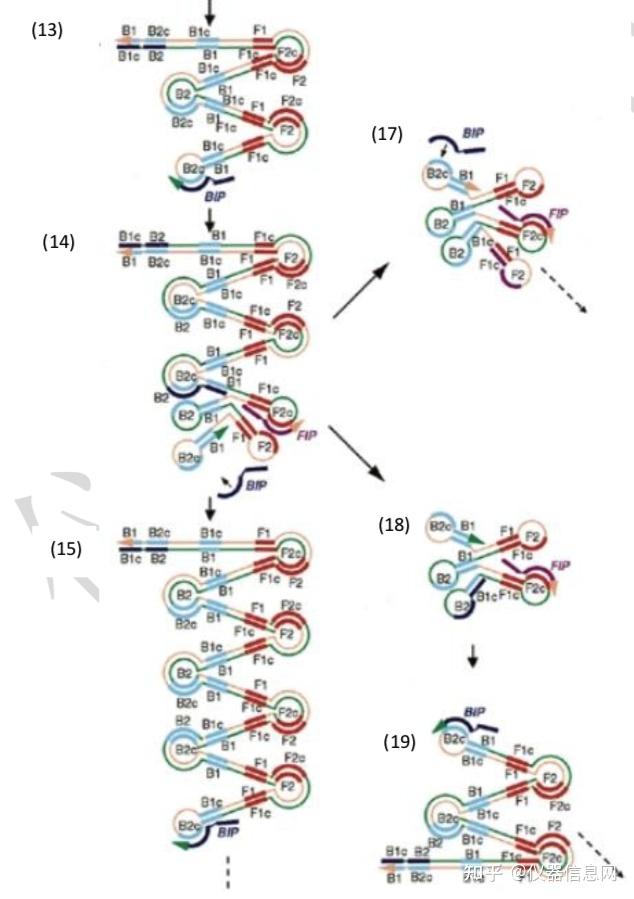
图 6.3 LAMP 延伸阶段示意图
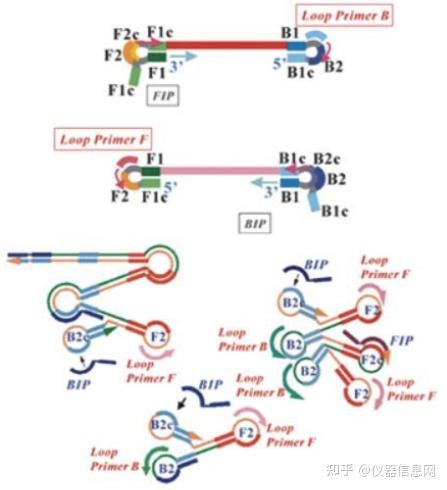
图 6.4 环引物在 LAMP 扩增中的作用示意图
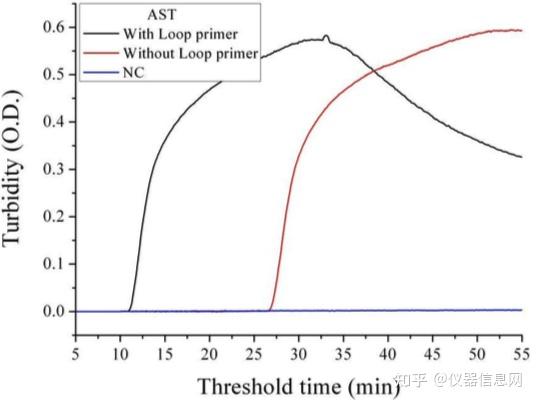
图 6.5 星状病毒环引物对 RT-LAMP 扩增的影响
第 2 节 环介导等温扩增(LAMP)引物设计
LAMP 引物设计基于靶序列上自 5’端开始依次标识为 F3、F2、F1、B1、B2、B3 的 6 个不同的区域(图 6.6),其中 F1、F2、F3 是靶基因上约 20bp 长的序列片段,B1、B2、B3 则是位于互补链上长度约 20bp 的序列,F1c 和 B1c 分别是 F1 和 B1 的互补区。4 条引物包 括上游外部引物 F3(Forward Outer Primer)、下游外部引物 B3(Backward Outer Primer)、 上游内部引物 FIP(Forward Inner Primer)和下游内部引物 BIP(Backward Inner Primer)。 各引物序列片段构成见图 6.6。此外,还可在 F1c 和 F2c 之间,或者 B1c 和 B2c 之间设计一 条或两条环引物 LF(Loop Primer Forward)、LB(Loop Primer Backward),促进 LAMP 扩 增效率。
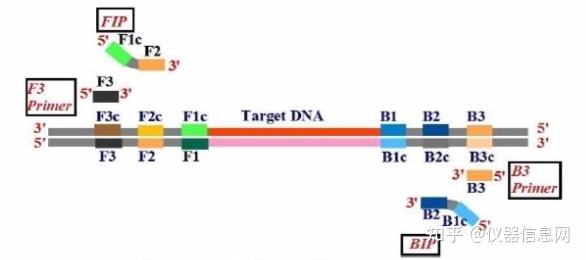
图 6.6 LAMP 引物示意图
LAMP 引物设计有 4 个关键影响因素:Tm 值、引物末端稳定性、GC 含量~和二级结构。 首先,Tm 值即 DNA 熔解温度。GC 含量占优势的序列 Tm 值在 60°C~65°C范围内,其中 F1c 和 B1c 要求 Tm 值 65°C(64°C~66°C),F2、F3、B2、B3 要求 Tm 值 60°C(59°C~61°C), 环引物 Tm 值 65°C(64°C~66°C)。反之,AT 含量占优势的序列 Tm 值则在 55°C~60°C范围 内。其次,引物的末端作为 DNA 合成的起始位点必须有一定程度的稳定性。F2/B2、F3/B3 和 LF/LB 的 3’端以及 F1c 和 B1c 的 5’端自由能应不大于-4kcal/mol。自由能变化(∆G)等 于引物与模板结合后产物自由能减去初始反应物自由能。引物与模板的结合是一个平衡反应, 只有当∆G 小于零时结合反应才会发生,且∆G 越小(负数的绝对值越大),引物越容易同模 板结合。此外,引物 GC 含量应在 40%至 65%之间,在 50%~60%范围内时为最佳。同时, 所设计的引物尤其是内引物不易形成二级结构也同样非常重要,为防止引物二聚体的形成, 需要确保引物 3’端不含较多的 AT 碱基,且与其他引物不存在互补序列。最后,各引物所识别的序列片段间应有一定合适的间距,如 F2 末端至 B2 末端(LAMP 扩增区域)间距 120~160 个碱基,F2 的 5’端至 F1 的 5’端(形成环结构的区域)间距 40~60 个碱基,F2 与 F3 间隔 0~60 个碱基(图 6.7)
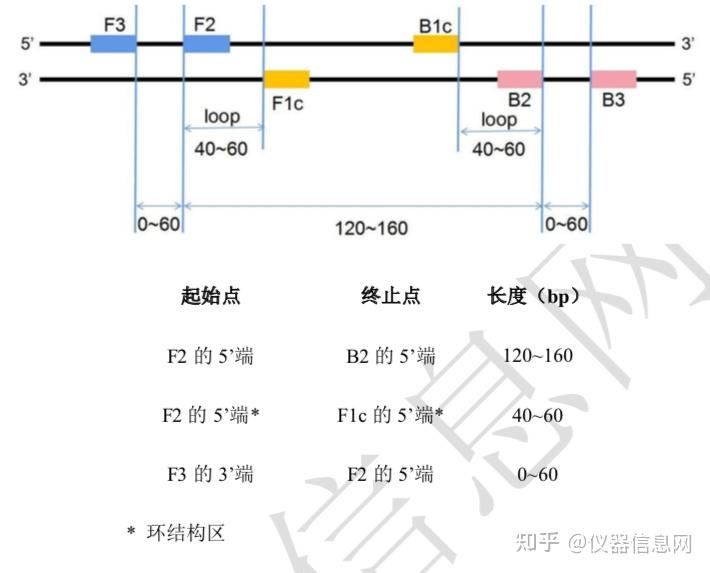
图 6.7 LAMP 引物间距
LAMP 引物设计流程:首先进行常规 LAMP 引物(FIP、BIP、F3 和 B3)的设计,经实 际扩增反应验证,结果满意的可以选做 LAMP 引物。如果没有出现有效扩增,或者扩增结 果不理想,则需要重新进行引物设计。进而利用所选择的 LAMP 常规引物序列进行环引物 的设计,同样需经实验验证,可有效提升扩增速率的才可选做环引物,否则需重新进行设计。 当然,环引物对于 LAMP 扩增不是必须的。
LAMP 引物设计可以使用在线设计软件 PrimerExplorer (http://primerexplorer.jp/e/)。软 件操作分为简易模式和专业模式两种。前者不需要改变引物设计参数,并自动缩小侯选引物 对范围,给出按优先级排序的可能产生有效扩增的 5 套候选引物。专业模式用于特定引物的 设计,用户可以改变引物设计参数,并可以规定所设计的引物对数量。通常对于一般序列(45 <GC% < 60)引物设计参数为默认设置,如果是 AT 富含序列(GC% < 45)或者 GC 富含序 (GC% > 60),则引物的设计参数应满足表 6.1 中的要求。

表 6.1 特殊靶序列的 LAMP 引物设计参数要求 59
当靶序列载入系统后,PrimerExplorer 会自动判断 GC 含量,并将靶序列按照 AT 富含 序列(GC% < 45)、一般序列(45 <GC% < 60)和 GC 富含序(GC% > 60)三种类型分别 匹配不同的引物设计参数。系统可以对设计出的引物末端序列进行自动检查,去除存在互补 序列(如 CCCGGG、GAATTC)或者多个相同核苷酸序列(如 CCGGGG、AATTTT)的引 物对。同时还会检查引物与靶序列的互补性,如果引物末端与扩增区以外的靶序列也存在互 补,则可能产生非特异性扩增,这样的候选引物也需去除。所设计的引物关键信息如引物在 靶序列中所在位置、Tm 值等可以自动保存并下载到 Excel 文件中。
下面以人星状病毒 1 型(Human astrovirus, HAstV)为例简要介绍 LAMP 引物设计的一 般过程。首先选择 HAstV 的衣壳蛋白编码基因(ORF2)作为靶序列(长度 1994 bp)。在 PrimerExplorer V5 的起始页面(图 6.8)点击“浏览”按钮,导入靶序列文件。靶序列长度应 小于 2kb,文件格式可以是 TXT(仅含序列)或 FASTA,也可以是 GenBank 格式。然后选 择引物设计条件参数,默认的参数设置为“Automatic Judgment”,该状态下会自动计算序列 的 GC 含量,并自动匹配合适的引物设计条件,点击“Primer Design”,进入引物设计页面(图 6.9)。点击“Generate”按钮,在右侧方框内即可显示有 5 对引物生成,继而点击“Display”按 钮,则进入引物列表页面(图 6.10)。可以点击“Save List”将引物列表信息保存到 Excel 文件 中。选中窗口左侧“Primer ID”对应的方框后点击“Confirm”,则出现引物对信息核查界面(图 6.11),根据引物对关键参数以及所扩增的靶序列是否符合预期从而选出最优引物对。在比 对分析引物性能时,需要特别关注 F2、B2 的 3’端,F1c、B1c 的 5’端,它们作为扩增的起 始位点,其稳定性非常重要。因此,代表稳定性的关键参数∆G 应小于等于-4.0kcal/mol,也 就是说末端∆G=-6.5kcal/mol 比末端∆G=-4.0kcal/mol 的引物更稳定。在每个引物对 ID 号的上 方,有一个“Primer Information”按钮,点击并保存文件以备用于后续环引物的设计。
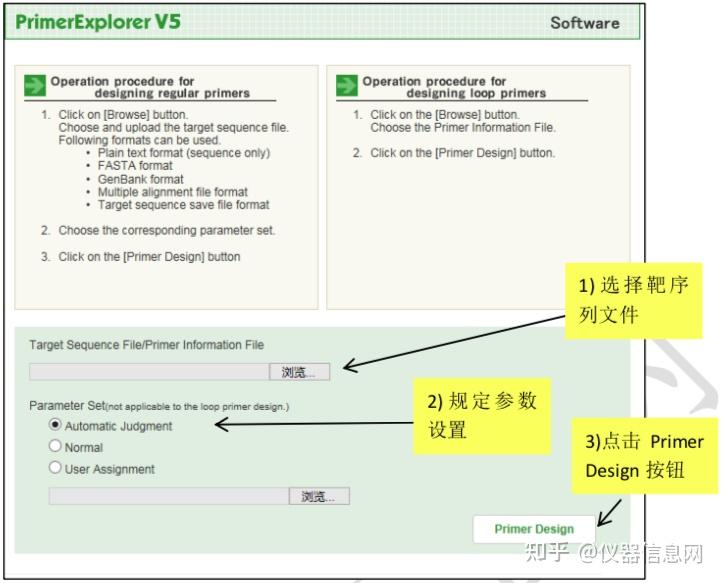
图 6.8 引物设计起始页面
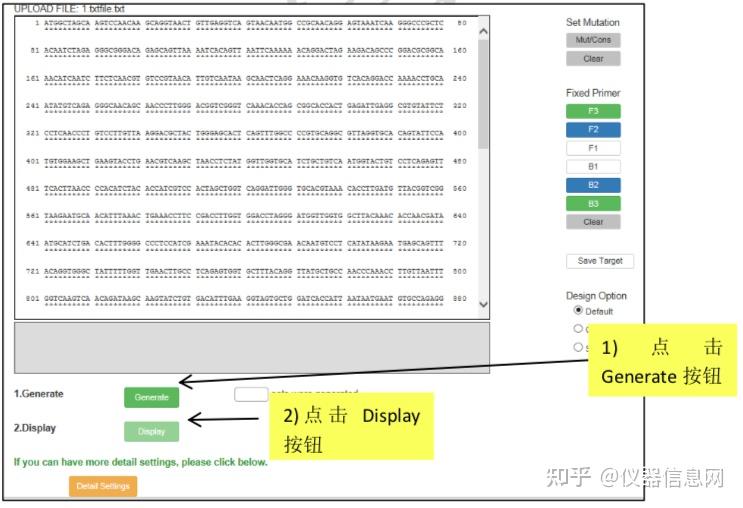
图 6.9 引物设计页面
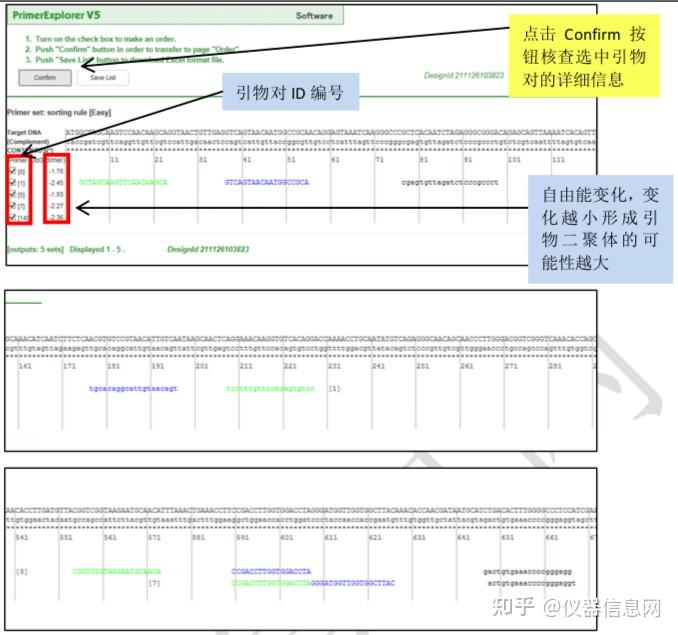
图 6.10 引物列表页面(部分) 绿色大写字母代表 F3 区,蓝色大写字母代表 F2 区,黑色小写字母代表 F1c 区,黑色大写 字母代表 B1c 区,蓝色小写字母代表 B2 区,绿色小写字母代表 B3 区
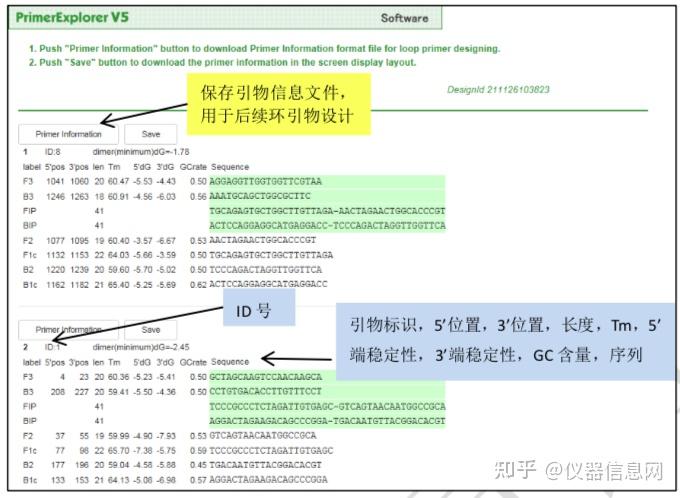
图 6.11 引物对信息核查界面
设计环引物时,回到引物设计的初始界面(图 6.8),点击“浏览”按钮后选择所保存的 “Primer Information”文件,然后点击“Primer Design”,进入环引物设计窗口(图 6.12),保持 默认参数设置,直接点击“Generate”按钮,显示有 4 套环引物生成,点击“Display”,进入环 引物列表界面(图 6.13),同样选中某环引物 ID 号左侧的方框,点击“Confirm”,进入环引 物信息核查界面(图 6.14),同样按照常规引物的筛选原则选出最优的环引物。
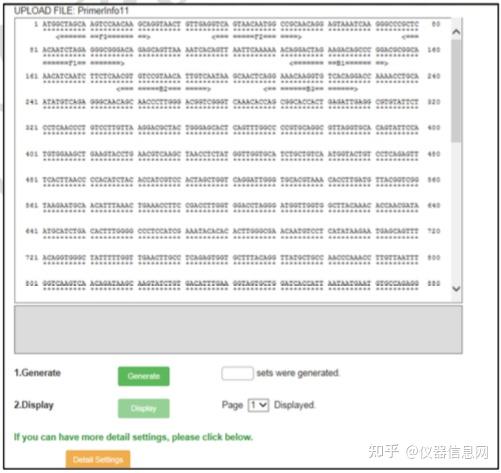
图 6.12 环引物设计界面
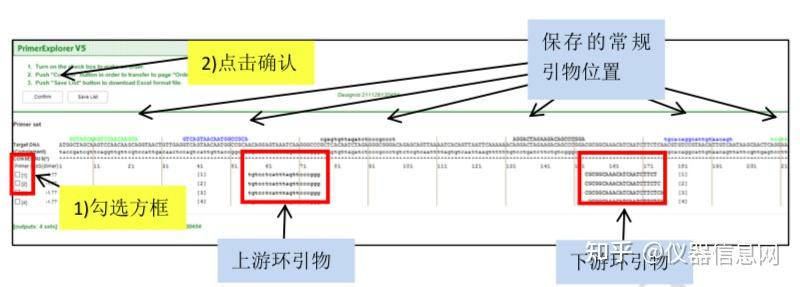
图 6.13 环引物列表界面
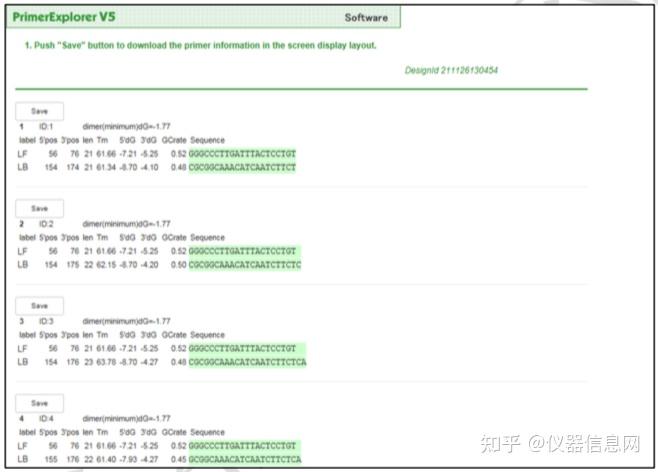
图 6.14 环引物信息核查界面
第 3 节 环介导等温扩增产物分析
LAMP 方法已被用于临床、食品和环境样本中多种病原的检测,如细菌、病毒、寄生虫 和原生动物等。其扩增产物的检测存在多种方式,且不断向更加准确、简便、高效的方向发展。
首先,LAMP 扩增产物可以通过琼脂糖凝胶电泳进行检测。LAMP 扩增后可以产生大 量具有不同长度、不同数目茎环结构和反向重复序列的 DNA 混合物,琼脂糖凝胶电泳呈现 瀑布状梯形条带。针对扩增产物中所包含的特异性酶切位点进行酶切,还可以对扩增产物的特异性进行验证。例如 Shirato K 等人建立的呼吸道合胞病毒(respiratory syncytial virus,RSV) 64 RT-LAMP 检测方法中,分别利用限制性内切酶 NlaIII和 XbaI对 A 亚型扩增产物和 B 亚型 扩增产物进行酶切,37°C作用 1h 后电泳检测,特异性扩增产物会由原先的瀑布状梯形条带 变为清晰可数的几条电泳条带(图 6.15)。电泳检测的缺点是需要打开扩增管,高浓度 DNA 产物容易产生气溶胶造成实验室环境污染。
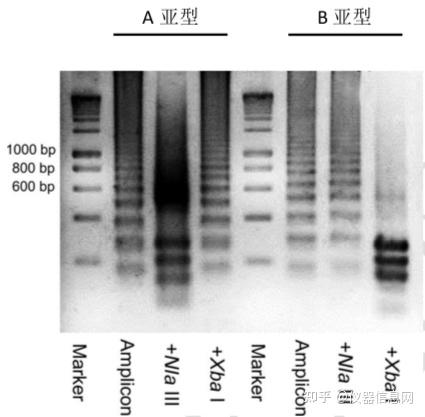
图 6.15 RSV LAMP 扩增产物酶切后琼脂糖凝胶电泳图
其次,Mori 等人于 2001 年提出了一种可以在密闭系统中通过浊度分析鉴定 LAMP 扩增 的方法。由于 LAMP 反应效率极高,在 15~60min 内便可使 DNA 的量放大 109~1010 倍,在 核酸大量合成时,从 dNTPs 析出的焦磷酸根离子与反应缓冲液中的镁离子结合,产生副产 物—焦磷酸镁,形成乳白色沉淀。而且焦磷酸镁的产量与扩增产物浓度存在一定相关性,因 此可在扩增的终末阶段肉眼观查乳白色沉淀判断扩增有效性,还可以通过浊度仪来实时监测 浊度的变化情况。利用实时浊度仪每隔 6s 记录一次反应液在 400nm 处的吸光值(OD400), 可以对反应管中的浊度变化进行动态监测,借以分析整个扩增反应的动态过程。一般将扩增 反应的阈值线设定在 0.1(大于阴性对照平均浊度值的 2 倍以上),浊度值超过 0.1 即认为是 阳性反应,而扩增过程中反应体系浊度达到阈值线时所对应的扩增时间称之为阈时间,以 Tt(Treshold time)表示。扩增的靶标分子含量越高,阳性信号出现的就越早,Tt 值就越小。 例如,RT-LAMP 对 10 倍梯度稀释的星状病毒 RNA 标准品进行检测,发现病毒 RNA 稀释 度越高,反应出现阳性信号所需要的时间(Tt)也越长,以病毒 RNA 浓度对数值为横坐标, 对应的阈时间为纵坐标绘图,发现星状病毒 RNA 在 102-107 拷贝数范围内模板量同阈时间存 在良好的线性关系,线性相关系数(R2)为 0.9905。类似于实时荧光 RT-PCR,这使得 RT-LAMP 也可以作为一种合适的定量工具,用于样品中病毒 RNA 拷贝数的定量分析。
然而,由于浊度信号是来源于核酸扩增的副产物,而不是引物特异性扩增的直接产物, 因此无法完全排除检测到非特异性扩增的可能。由宿主来源的近源 DNA 片段或引物二聚体 产生的非特异性 DNA 扩增都有可能带来浊度信号的增加,尽管这种情况很少见,还是应引 起足够的重视,特别是在建立 LAMP 检测方法时,应对引物的完整性和适用性做充分的验 证和比对分析。

图 6.16 LAMP 扩增产物目视检查混浊
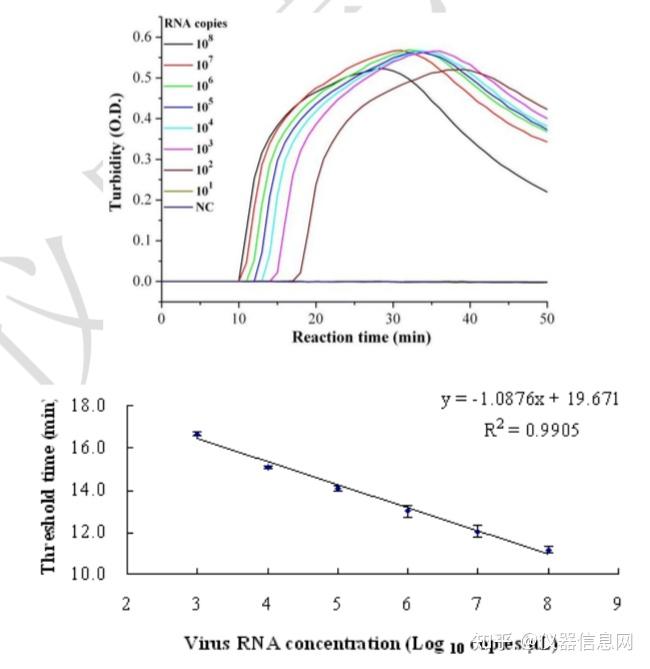
图 6.17 星状病毒 RT-LAMP 扩增实时浊度检测:浊度变化动态曲线(A)与扩增标准曲线 (B)
此外,染色检测法。根据染料对反应是否产生抑制,染料可在反应后或反应前添加。但 在反应后开盖添加,易在空气中产生气溶胶,并在之后的检测中造成假阳性。染料和辅助剂 的种类都会对反应效率及结果判断造成影响。因此,根据染料、辅助剂的作用特点进行合理 的选择及应用,对 LAMP 结果的正确性和可靠性有重要意义。染料的种类有很多,应用较 多的有 3 种:钙黄绿素、SYBR Green 和羟基萘酚蓝 ( 表 6.2 ) 。

SYBR Green I 是一种荧光染料,反应结果阳性时体系呈绿色,阴性体系呈橙色,肉眼 就可观察反应结果,也可置于紫外灯下观察反应结果。但荧光染料的使用具有两面性,使用 不同的核酸染料会对 LAMP 反应造成一定的影响。若是反应前加入,由于荧光染料对 DNA 分子的高结合力和亲和力(因为荧光染料会与双链 DNA 的小沟部分结合,一旦结合其荧光 信号会增强 800~1000 倍),从而阻碍 Bst DNA 聚合酶链置换活性。反应结束后开盖加入, 存在气溶胶污染,从而造成假阳性。不少试验人员选择反应前将 SYBR Green I 滴加于反应 管的盖上,反应结束后,将染料弹入或离心与反应体系混匀,这种操作方式避免了对反应效 率的影响以及产生气溶胶污染。但需注意的是,在 PCR 仪开启热盖的模式下,染料会变干 凝固,不易回溶,所以宜采用水浴或 PCR 仪不开热盖的模式。使用 SYBR Green I 并不能做 到特异性的显示扩增产物,引物二聚体或非特异性扩增产物也能结合荧光染料显色,易造成 假阳性,对引物的设计要求较严格。
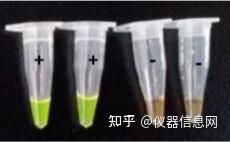
图 6.18 LAMP 法 SYBR Green I 染色结果
钙黄绿素( Calcein) 是一种络合指示剂和荧光指示剂。Tomita 等用钙黄绿素和氯化锰开 发了一种有效的 LAMP 终点检测方法。将钙黄绿素溶于二甲基亚砜( DMSO) 成 5 mmol /L 溶液,再用蒸馏水配制成 0.5mmol /L 钙黄绿素、10 mmol /L MnCl2 的储备溶液。钙黄绿素 与 Mn2+结合时处于荧光淬灭状态,当随着大量 DNA 双链的合成,反应体系当中产生的焦磷 酸根离子副产物,使得 Mn2+与磷酸根结合生成沉淀解除淬灭状态,钙黄绿素与 Mg2+结合, 又产生荧光。但在 2010 年,Wastling 等发现,与没有添加指示剂的反应相比,加入钙黄绿 素似乎降低了 LAMP 的敏感性。实际上,钙绿黄素的使用对反应体系当中的 Mg2+的浓度要 求很高,有一定的偏差,都会造成反应结果误差,在低浓度的 Mg2+条件下,酶活性降低, 产物量下降,在 Mg2+浓度过高时,会出现非特异性反应,所以应选择合适的 Mg2+浓度。钙 黄绿素在一定程度上抑制了 LAMP 反应,另一个原因是钙黄绿素和双链 DNA 之间的相互 作用导致了反应灵敏度的降低。Loopamp 荧光检测试剂(LMP221) 的主要成分为钙黄绿素, 并就其对紫外线的照射要求进行了研究,发现: 当钙黄绿素处于短波长(240 nm)到长波长 (370 nm)时,产生黄绿色荧光; 在中间波长(325 nm)的激发光下荧光最强; 当激发光接 近 320 nm 时,虽然从阳性样品观察到的荧光增强,但阴性对照的荧光也加强。因此,建议 在短波长(240~260 nm)或长波长(350~370 nm)的激发光下进行 LAMP 可视荧光检测, 通过比较样品与阳性、阴性对照的荧光强度进行判断。

图 6.19 LAMP 法钙黄绿素染料紫外灯下显色结果
羟基萘酚蓝(hydroxy naphthol blue,HNB) 也是一种金属离子指示剂,可与反应体系当中的 Mg2+结合而成紫罗兰色,当 DNA 双链合成后,生成焦磷酸镁,失去镁离子的 HNB 就变成了天蓝色。HNB 在终浓度为 120 μmol /L 时对扩增没有抑制,可在反应前加入。但是 该种染料在紫外灯下颜色区分不明显,且无成品化试剂,进行反应时需要按照一定浓度现用 现配,且 HNB 不稳定,不容易保存,使用不便。另一个问题是 HNB 的颜色反应肉眼区别 不明显,不容易察觉颜色变化,虽然目前很多研究者在使用本方法,但并没有获得大家的认 可。
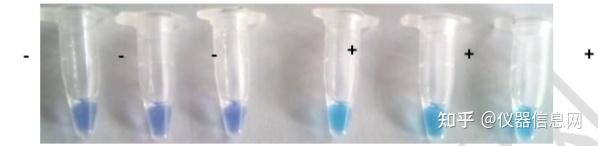
图 6.20 LAMP 法 HNB 染色结果
上述 3 种染料相互比较,SYBR Green I 和 HNB 的检测灵敏度比钙黄绿素高 10 倍,可 能由于 Mn2+的抑制,或者钙黄绿素与 DNA 的相互作用引起钙黄绿素灵敏度的降低。
最后,还可通过恒温扩增微流控芯片实时观察反应结果。微全分析系统(Miniaturized Total Analysis Systems,TAS)或称芯片实验室(Laboratory-on-a-Chip,简称 LOC)是跨学 科的新领域,目标是通过分析化学、微机电加工(MEMS)、计算机、电子学、材料科学及 生物学、医学的交叉,实现化学分析系统从试样处理到检测的整体微型化、自动化、集成化 与便携化。微流控分析(Microfluidic Analysis)是微全分析系统的主要组成部分, 而将化 学分析的多种功能集成在邮票大小的芯片上的微流控芯片(Microfluidic chips)又是当前最 活跃的发展前沿,代表着 21 世纪分析仪器走向微型化、集成化的发展方向。具有微小可控、 功能集成的微流控芯片, 能够简化操作过程、加快分析速度,还可避免常规方法中样品转 移所带来的损失和污染, 为病原微生物等的分析提供了一个新的平台, 对于发展强大的生 物分子床边快速诊断(Point of care testing,POCT)具有非常重大的意义。
基于微流控芯片的核酸扩增检测系统(Nucleic acid amplification test,NAAT)是微流控 技术最有前景的应用之一,它简化了核酸扩增检测中繁琐的样品前处理和扩增产物检测步骤。 在多种 NAAT 检测方法中,LAMP 与微流控技术结合的核酸扩增检测系统则最有潜力被应 用在 POCT 上。近年来已有不少研究报道将环介导等温扩增与微流控芯片结合,用于检测 病原微生物、癌症生物标志物及其他靶基因。该方法具有特异性强、敏感度高、耗样量少、 耗时短、检测效率高、操作简便等诸多优点。整合环介导等温核酸扩增和微流控芯片技术,建立单重/多重、定性/定量 LAMP 微流控芯片模块,发展快速、灵敏、特异和适合床边检测 的生物分子分析技术,将为我国食品安全、重大传染病防治、肿瘤及遗传性疾病等的快速准 确诊断提供强有力的技术保障,为创新技术的发展提供良好的示范和引导。
第七章 数字PCR
数字 PCR(Digital PCR,dPCR)作为第三代 PCR 技术,是一种全新的核酸检测和定量 的方法。自 1999 年 Vogelstein 和 Kinzler 提出了 dPCR 的概念以来,相关技术和产业化发展 都非常迅速,已被成功用于临床诊断、转基因成分定量、单细胞基因表达、环境微生物检测 等领域。
第 1 节 数字PCR原理
dPCR 技术一般包括两部分内容,即 PCR 扩增和荧光信号分析。在 PCR 扩增阶段,dPCR 是将 qPCR 体系等量均分到相互隔离的大数量(多数大于 10000 个)微小的芯片微孔或微 滴液珠等反应单元中。理想状态下一个反应体系中至多包含一个待测核酸分子。dPCR 的反 应过程与 qPCR 相似.在荧光信号分析阶段,采用终端检测,是对每个反应单元的荧光信号 进行采集,扩增后的核酸分子在荧光下显示荧光信号,表示为“1”,或没有荧光信号,表示 为“0” ,通过泊松分布对荧光信号“0”和“1” 进行统计分析,即可实现对核酸含量的测定。
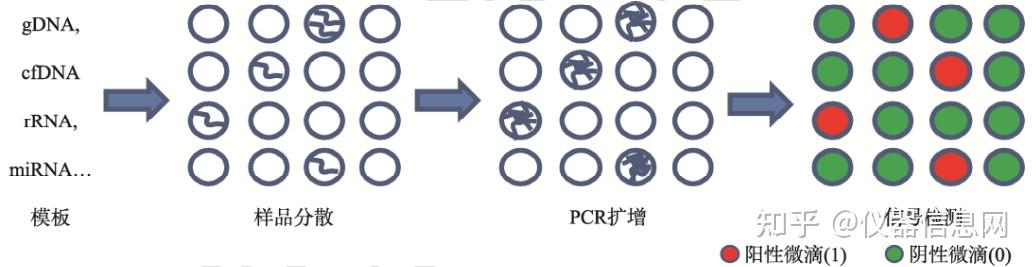
图 7.1 数字 PCR 的作用原理
在 dPCR 统计分析中引入了泊松统计方法进行数据的校准。 泊松分布是一种离散概 率分布,是指在固定的时间或空间内发生给定事件的概率,假定事件以固定的平均速率发生, 并与上一次以来的事件无关。泊松概率分布公式如下:
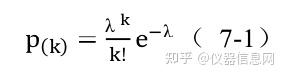
其中 p(k)是确定观察到 k 目标分子的概率,λ是每个反应单元内平均目标数,k 是反应 单元内实际存在目标分子数。dPCR 结果分析是基于以下假设:即目标序列在反应单元内的 分布是近乎完美的泊松过程。设定 P 为阳性反应单元数目,N 为总反应单元数目,VP 为 单个反应单元体积,D 为模板稀释因子,每微升样品中 DNA 拷贝数计算公式见如下:
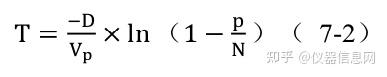
置信区间(confidence interval)是指由样本统计量所构造的总体参数的估计区间。在统计 学中,一个概率样本的置信区间是对这个样本的某个总体参数的区间估计。在各平台的仪器 设置中,一般选择泊松分布 95%的置信区间计算误差线。
dPCR 的关键是稀释模版,通过将一个样本分成几十个到几万份,分配到不同的反应单 元进行 PCR 扩增。dPCR 标准反应体系分配的过程(稀释过程)能够极大的降低与目标序列 有竞争性作用的背景序列浓度,因此 dPCR 特别适合在复杂背景中检测稀有突变。同时,dPCR 极高的灵敏度使得在检测极低浓度的样品时不需要进行预富集工作。dPCR 相比较于 qPCR 具备了以下优势:1)无需依赖有证标准物质或其他标准品,无需内参基因,不必绘制标准 曲线;2)具有更高的精确度、灵敏性和重复性,易于标准化;3)更适合低拷贝数的核酸检 测;4)由于 ddPCR 是终点检测,对抑制剂的耐受度更高,从而可降低由食品基质所导致的 检测偏差。
dPCR 引物探针的设计原则同 qPCR 基本一致。PCR 产物的特异性取决于引物与模板 DNA 特异性识别与结合的程度,因此引物是 PCR 反应特异性的关键。表 7.1 列出引物设计 的关键特性优化原则,现今有多种计算机程序可以对引物进行设计、选择和优化。dPCR 普 遍使用 TaqMan 探针或荧光染料作为荧光信号产生的方式。根据商业化仪器的不同要求合理 选择使用荧光染料或 TaqMan 探针,比如 QX100(Bio-Rad)仅可以使用 TaqMan 探针作为 荧光信号的产生方式,QX200 及其他种类的商业化数字 PCR 两者均可使用。表 7.2 则给出 了 TaqMan 探针设计的关键点。


表 7.1 引物特异性及优化设计

表 7.2TaqMan 探针设计
第 2 节 数字PCR应用特点
自从 1999 年数字 PCR 技术概念提出以来,科学家们就期望通过样本分散的方式实现 绝对定量和低丰度样本检测,该技术近些年获得了突飞猛进的发展。数字 PCR 与实时荧光 定量 PCR 的不同之处在于其将样本中的核酸分子在若干独立微反应单元中随机分散和检 测,这种策略使核酸绝对定量成为可能。
众多研究者对数字 PCR 和实时荧光定量 PCR 做了对比,其优缺点归纳总结为以下几 个方面:1就目前市场上已有的数字 PCR 没备来看,实时荧光定量 PCR 在检测的线性范 围上更具优势,这意味着同一个反应体系内可以对各种表达丰度的样品进行分析。目前实时 荧光定量 PCR 已经能实现高通量样本条件下的自动化操作2数字 PCR 可以实现单分子层 面上的绝对定量,彻底摆脱对标准曲线的依赖而直接给出靶序列的拷贝数,提高了实验结 果在批内和批间的稳定性,甚至能用标准品来定标;3基于绝对定量的方式,数字 PCR 结 果的高重复性和高精度可实现微小差异的基因表达分析;4同等条件下,数字 PCR 在检测 灵敏度方面更具优势。
总之,全新的数字化检测方式和分子计数的定量手段,赋予了数字技术诸多优点,即 无需标准曲线和参照,对影响效率的抑制物不敏感,大大提高了检测灵敏度、精确度、准 确度和重复性,实现了真正意义上的绝对定量。本章从临床医学、食品安全检测领域展开介绍。
1、数字 PCR 技术在临床医学中的应用
(1)肿瘤诊疗
数字 PCR 具有较高的检测灵敏度,因此除了将常规的活检组织作为基因检测对象外, 还可以与液态活检技术相结合,达到对核酸含量较微小的样本进行基因检测的目的。如以 血液中来自凋亡或坏死的肿瘤细胞的 DNA 片段为检测对象的游离 DNA( cell-free DNA, cfDNA) 、循环肿瘤 DNA( circulatingtumor DNA,ctDNA) ,代表了肿瘤的基因组信息, 可以用于解释肿瘤发生机制,反映肿瘤发展状况。非编码 RNA 也可以提供额外的生物标 志物信息用于反映相关肿瘤的生长状态。除血浆、血清等主要核酸来源外,脑脊液、尿液、 痰液、胸腔积液、腹水、粪便等样本也可提供 DNA 和 RNA 以供数字 PCR 分析。Day 等 对数字 PCR 超高的突变检测灵敏度在突变等位基因频率(MAF)检测方面的应用进行了研究和分析。对于每一种肺癌中常见的 EGFR 突变,仅含有 L858R 突变而不含 T90M 突变 的肿瘤组织,传统 sanger 测序(灵敏度为 20%~30%)即可检测出该突变;肿瘤组织常出 现含有 T90M 突变的局部细胞克隆,这样的稀有突变需要灵敏度更高的检测手段方能检出; 另一种可能出现的情况,肿瘤中存在 3 种局部细胞克隆,但占优势的正常细胞克隆不含有 任何 EGFR 突变。当前针对上述 3 种情况的癌症普遍采用相同的靶向治疗方案,但是这 3 种肺癌对 EGFR TKIS 靶向药物的反应却有很大差异。数字 PCR 技术结合“液体活检”的理 念就可以实现对肿瘤的实时动态基因分型,从而为个体化用药和预后提供必要信息。
(2)无创产前检查
产前检查是减少出生缺陷二级预防措施的重要手段,其目标是预防严重缺陷儿的出生, 重要性不言而喻。研究人员把 ddPCR 用于非侵入性产前诊断,包括胎儿染色体非整倍体检 测或胎儿 RhD 基因分型。数字 PCR 能以导致遗传疾病的异常基因为靶标,只需抽取少量 母体外周血提取总核酸,克服母体 DNA 背景的干扰,运用多种策略对其中含量极低的游 离胎儿 DNA/RNA 标记进行精确定量检测分析,即可达到在基因水平对各种遗传疾病如脊 髓性肌萎缩症(SMA)准确检测。
Zhong 等提出了一种基于数字 PCR 技术来进行基因拷贝数分析的新方法,并初步应用 于SMA患者的诊断研究,实现了对SMN基因拷贝数的精确定量。 Stabley等应用数字PCR 技术对 SMA 患者的 SMN1 和 SMN2 基因拷贝数进行了精确检测。结果表明,数字 PCR 技 术可以实现 0~3 拷贝的 SMN 基因和 0~5 拷贝的 SMN2 基因的准确定量;数字 PCR 检测 SMN1 和 SMN2 基因拷贝数的 CV 值分别为 1.6%~3.7%和 2.1%~3.7%,相对应的 TaqMan qPCR 技术检测的 CV 值分别为 5.2%~9.7%和 0.8%~9.7%,显示出数字 PCR 技术在 SMA 分 子诊断方面具有很大优势。
(3)病原微生物检测和传染性疾病防控
ddPCR 采用绝对定量的手段,无需标准品作为参照,有助于确定病原体载量,可用于 目标中多种序列的定量和含抑制剂样品的定量。在缺少标准品无法进行 qPCR 扩增的情况 下,ddPCR 可用于检测各种临床样本中的 DNA 和 RNA 病毒拷贝数。已有研究表明 ddPCR 可定量分析多种病毒,包括艾滋病毒 DNA 和 RNA,巨细胞病毒,乙型肝炎病毒,人类乳 头状瘤病毒,人类 t 细胞白血病病毒,人类鼻病毒,丙型肝炎病毒(HCV),E 型肝炎病毒
等。细菌和原生动物从呼吸道、食道、生殖道等进入人体,细菌性传染病有时很难被识别,因为特定病原体具有特定的生物学阶段,在这个阶段,为躲避免疫系统的清洗,病原体的 浓度非常低。由于 ddPCR 对低丰度 DNA 具有良好的敏感性,因此研究人员在细菌性传染 病的病原体检测方面进行了尝试。有研究表明 ddPCR 可定量检测结核分枝杆菌和幽门螺杆 菌等细菌和疟疾寄生虫等。
HCV 感染后的临床症状温和,但是 20%左右的感染者会罹患慢性肝炎、肝硬化,甚至 发展成肝癌。作为一种 RNA 病毒,HCV 具有显著异源性和高度可变性的特点。HCV 核心蛋 白序列第 70 氨基酸的突变与肝病演变相关,并且是一个独立的肝癌风险评估因子。直接测 序通常可以用于检测突变,但是当测序数据有重叠时,很难判定突变的存在。这极大的制约 了人们对 HCV 基因组突变与肝病发展关系的研究。有研究人员建立了基于 QX100 液滴式 数字 PCR 的 HCV aa70 突变检测技术,并对其进行了系统的验证评价,同时与直接测序和 罗氏 COBAS TaqManHCV RNA qPCR 检测试剂盒做了对比。在存在高浓度野生型背景时, 数字 PCR 对 HCV aa70 突变的检出限可达 0.005%,阳性检出率远高于测序技术;数字 PCR 重复性也明显优于罗氏 qPCR-HCV 检测试剂盒。数据显示,QX100 液滴式数字 PCR 不仅 可以分析 HCV 核心蛋白 aa70 突变率,还能对病毒多态性基因组中靶基因的突变进行精确 定量,成为病毒学研究的有力技术手段。
结核病(TB)是一种由结核分枝杆菌(MTB)引起的影响肺部的传染病。它在早期感染时 没有症状,被称为潜伏性结核病。大约 10%的潜伏性感染会发展为活动性疾病,如果不加 以治疗,有 50%的死亡率。传统的血培养或抗酸杆菌的多次痰培养通常需要 2 ~ 6 周,因 此,用 PCR 方法定量检测结核分枝杆菌 DNA 是一种可靠、可重复性强的结核病诊断方法。 PCR 近年来正在成为肺结核(PTB)的一种诊断工具。与 qPCR 不同的是,ddPCR 可以提供 样品中细菌数量的直接计数,从而提高检测结核的灵敏度。研究表明,ddPCR 能较好地检 测人血浆中的少量细胞游离 DNA 靶点。一些特殊患者(如无呼吸道病变的外肺结核患者、 播散性肺结核患者、婴儿或儿童)难以获得足够的标本,这些病例过去只能通过支气管镜 检查或手术等侵入性方法获取样本进行痰培养筛查,现在可以通过 ddPCR 检测结核患者血 液样本中的 mtb 特异性 DNA 片段。随着 ddPCR 技术的发展和性能的提高,未来可以更快 速、准确地诊断结核病。
2、数字 PCR 技术在食品安全检测中的应用
目前在食品安全领域用实时荧光定量 PCR 实现的检测基本都可以利用数字 PCR 完成, 特别是在转基因(genetically modified organism,GMO)成分、动植物源性检测和食源性 微生物检测等领域被广泛应用,并形成了一系列国家标准和行业标准。现行有效的国家标 准有 GB/T 33526-2017 转基因植物产品数字 PCR 检测方法、GB/T 38132-2019 转基因植物 品系定量检测数字 PCR 法等。现行有效的行业标准有 SN/T 5325-2020 出口食品中食源性 病毒定量检测 数字 PCR 法 7 个系列标准;2022 年 6 月 1 号还有 SN/T 5364-2021 出口食 品中致病菌检测方法 微滴式数字 PCR 法 8 个系列行业标准即将实施。
(1)转基因成分检测
目前国内外对转基因作物及其相关制品的检测主要是基于外源蛋白靶标和外源核酸 成分的检测来展开的,建立了多种快速、灵敏的转基因定性和定量检测方法,这些方法通 常分为两组:基于蛋白质的方法和基于 DNA 的方法。由于一些性能参数,目前在转基因 检测中应用最多的还是基于 DNA 序列的 PCR 检测方法。数字 PCR 作为更精确和更灵敏的 DNA 定量检测新技术,实现了单分子 DNA 的绝对定量,为转基因产品的定量分析提供了 新的技术手段。
在转基因生物研发与安全评价过程中,外源基因整合进入受体基因组的方式(位置拷 贝数和旁侧序列等)会影响目的基因和蛋白的表达及外源基因的遗传稳定性,特别是整合 的拷贝数的影响较大。因此,转基因生物外源基因整合的拷贝数分析是食品安全检测的关键 步骤之一,同时也是生物安全的重要评价参数。数字 PCR 不依赖标准品的绝对定量方式能 改变 GMO 检测对标准物质的依赖问题。转基因样本包括经过各种工艺制作的食品材料成 分复杂,其中含有大量 PCR 抑制物,而数字 PCR 对 PCR 效率不敏感,对抑制物耐受度高 的特性可以很好的解决这个问题。
Corbisier 等利用数字 PCR 分析了提取于 MON810 玉米种子 DNA 中外源基因和内标 准hmg基因的拷贝数之比,该结果与以质粒DNA 为标准物质进行qPCR检测的结果一致, 证明了数字 PCR 技术定量检测的可靠性,可以用来测量转基因相关 DNA 拷贝数比率。
对于食品及饲料中的转基因成分一般是通过实时荧光定量 PCR 技术进行测定的,但实 时荧光定量 PCR 对于痕量 DNA 的定量检测结果并不理想。Dany Morisset 等利用数字 PCR 对 MON810 转入基因及玉米 hng 基因(作为内参基因)进行了绝对定量,精度可达 5 拷贝, 动态范围超过 4 个数量级。结果表明数字 PCR 在检测低丰度 DNA 时具有更好的可重复性,
且对抑制因子更具数耐受性,数字 PCR 技术有望成为定量分析食品和饲料中转基因成分或其他物质的常规方法。
(2)食源性微生物检测
食源性致病微生物是影响食品安全的重要因素之一,如金黄色葡萄球菌、沙门氏菌、 单核细胞增生李斯特氏菌、大肠埃希氏菌、副溶血性弧菌等。这些微生物广泛存在于肉制 品、奶制品、水产品、蔬菜等多种食品中,是引起食物中毒的主要致病微生物。尽管食品 加工过程中对微生物污染严格控制,但仍无法完全避免食品污染引起的疾病或食品腐败, 食品污染致病微生物会影响消费者健康,同时也为企业造成较大经济损失。因此,对食品 原料、加工环境及最终产品中可能存在的微生物进行有效检测至关重要,对确保食品安全 性及保障食品品质具有重要意义。
近年来,随着社会对食品安全重视度的提高,相比定性检测,重要食源性致病菌定量 检测或是一个趋势。致病菌定量检测对于执行食品微生物限量标准、对食品中的致病微生 物进行风险评估更具有实际的意义,建立其快速、灵敏、特异的定量检测方法是控制食源 性疾病的关键。传统培养法是微生物定量检测的主要技术手段, 但需繁琐的增菌、分离、 鉴定等步骤, 费时费力,辅助的 qPCR 方法,虽在时间上有了很大突破, 但其结果的准确 性依赖于标准曲线的构建和引物的扩增效率,而 ddPCR 技术弥补了这一缺陷。同时由于 其抗抑制能力使其可能成为高通量筛选微生物以评估食品质量和安全性的有用手段。
除致病微生物外,酸奶、玫瑰酱等发酵食品中含有或添加大量多种益生菌。益生菌食 品功效的发挥主要取决于活菌的作用,因此快速准确检测该类食品中益生菌的活菌数量是 检测重心。随着研究技术的发展,针对益生菌种和株水平上性质差异的探讨也持续深入。 采用数字 PCR 技术可对其亚种进行鉴别并测定含量。
Roman Netzer 等利用数字 PCR 技术对水产养殖中重点细菌(包括单核细胞增生李斯特 氏菌)进行绝对定量。赵丽青等基于单核细胞增生李斯特氏菌 hlyA 基因建立了 ddPCR 检 测方法,得到计算公式为:初始浓度(CFU/g)=检测数据(copies/μL)×25,检出限可以达 到 90 CFU/mL,灵敏度非常高且重复性好,可高效、准确检测食品中核细胞增生李斯特氏 菌。赵新等根据沙门氏菌 invA 毒力基因序列对沙门氏菌进行快速检测并与传统培养法进行 对照验证,检测灵敏度可达到 102 CFU/mL,模拟污染沙门氏菌的金针菇样品实测与传统国 标法对比符合率 100%,可以在保障准确性的同时节省大量时间。Wang 等在其研究中也证明了数字 PCR 比 qPCR 节约 2h 左右预培养时间,且最低检出限可达到 102 CFU/mL。周巍 等根据金黄色葡萄球菌 nuc 基因设计特异性检测体系,利用 ddPCR 方法对发酵乳中金黄色 葡萄球菌 nuc 基因的检测特异性和灵敏度进行实验,检测限可达到 3.3×101 CFU/g,定量检 测结果精确,特异性好。魏咏新等建立的大肠埃希氏菌 O157:H7 的 ddPCR 检测技术的最 低定量限为 4.2 拷贝/反应,人工污染食品样品的检测限为 110 CFU/g,适用于食品中高污 染大肠埃希氏菌 O157:H7 的定量检测应用,为大肠埃希氏菌 O157:H7 的防控和监管工作提 供技术支持。
针对食品中益生菌的数字 PCR 定量研究比较少见,目前已有研究人员开展乳制品中多 种乳酸菌 ddPCR 检测方法的建立及标准制定,所建立的乳制品中植物乳酸菌、两双歧杆菌、 青春双歧杆菌 ddPCR 检测方法均具有良好的特异性,重复性,定量检测结果与 GB 4789.35 计数结果无显著统计学差异,可用于添加该乳酸菌的乳制品中乳酸菌的快速定量检测。
(3)动植物源性检测
由于市场需求扩大和原料价格上涨,部分企业或商家为减少成本获得高利润,加工过 程中在食品中掺入配料表中未经标识的成分,以假充真、以廉充贵。除此之外,在食品生 产加工过程中由于共用仓储和加工设备,造成产品无意掺杂未经标识的源性成分也是非常 普遍的现象。准确区别上述两种不同性质的源性成分掺入,直接关系到能否对食品掺假进行 有效监管。因此对于食品源性成分精准定量检测需求与日俱增。目前常用检测方法主要为 qPCR 法,该方法依赖标准曲线且易被抑制剂影响,现有研究表明,在建立源性成分精准 定量检测技术区分食品有意掺假和无意掺杂方面,qPCR 无法提供有效的解决办法。数字 PCR 技术在检测过程中直接获得源性成分的拷贝数,并依据拷贝数含量与质量的相关性, 估算源性成分的质量百分比,从而可以将有意掺假和无意掺杂区分开来。
数字 PCR 已经应用于猪、牛、羊、马、鸡、鸭、鹅等动物源性成分及核桃、大豆、椰 子、花生、红薯等植物源性成分的定量检测研究中。 CaiY等研究发现,烘干的未经加工 的生肉质量和 DNA 的质量、DNA 的质量和 DNA 的拷贝数检测数据都接近于线性关系。实 验表明可通过测定肉制食品中特定物种拷贝数含量为区分有意掺假和无意掺杂提供依据。 王珊等建立了一种定量检测羊肉制品中羊源性成分与猪源性成分的方法,并利用该方法对 市售的 3 份羊肉制品进行 qPCR 和数字 PCR 检测。用 qPCR 可以同时在 3 份样品中检测出 羊成分和猪成分,但只能粗略的从 Ct 值上判断其 DNA 含量的多少,根本无法准确定量样 品中羊源和猪源性成分的含量;数字 PCR 方法能检测出样品中的羊源及猪源性成分的 DNA 拷贝数,检测的猪源性成分含量相比较羊源性成分来讲,只能判定为是加工环节或者样品 流通中出现的无意沾染,而非故意添加。杨硕等建立了市售核桃乳中核桃源及主要掺杂物 种大豆 2 种源性成分的准确、快速多重 ddPCR 检测方法,该方法灵敏度高,核桃中掺杂 大豆的质量检测限为 0.5%,相对误差为 5.6%,用实际样品进行验证,样品大豆与核桃质 量之比高于 10%时,判断存在掺杂使假,大豆与核桃质量之比低于 0.2 时,极低的检出量 推断为工艺沾染,该研究建立的准确、快速的多重 ddPCR 定量检测方法可以作为鉴别核 桃乳中掺杂使假的有效手段。
通过对两种方法的比较发现,数字 PCR 方法可以对样品中,源性成分的含量比例进行 准确定量,这方便了食品检测部门对样品中某种源性成分的真伪做出判定。应用数字 PCR 方法定量检测食品中的源性成分是值得多考虑的行业标准发展方向,可以指导我国食品监 管部门对食品安全进行把关。
第 3 节 不同品牌数字PCR之间的区别
根据样品分散方式的不同,市场上数字 PCR 仪可分为两大类:(1)基于微流控芯片数 字 PCR 仪。此类平台又分为封闭式(美国 Fluidigm 公司 BioMark)和开放式(美国 ThermoFisher 公司 Quant Studio) 两种平台。封闭式平台是在硅片或石英玻璃上光刻多个 微管或微腔室,通过不同的阀门控制溶液的流动,实现样品制备、反应、分离和检测;开放 式平台是以超高密度亲疏水微孔芯片作为反应载体,芯片表面被处理成疏水,而微孔内部为 亲水,这种亲疏水结合的方式可使样品及反应体系轻易进入微孔而不会停留在表面,从而形 成密集的独立微反应室并避免了各反应之间的交叉污染。封闭式和开放式平台的仪器系统可 以称之为“固-液相”数字 PCR 系统。(2)基于油包水技术的微滴数字 PCR 平台(美国 Bio-Rad 公司 QX100、QX200 和 Raindance 公司的 RainDrop 系列、中国瑞讯生物公司 DropX-2000 以及法国 Stilla Technologies 公司 Naica System)。该平台通过液滴发生器将乳 化液滴分散成大小均一的液滴反应单元,这些仪器系统则可以称为“液-液相”数字 PCR 系统。
“固-液相”数字 PCR 技术由于采用预加工的区间进行样品分隔,因此各反应单元相互独立,分隔明确,不易出现相互串扰,反应单元的均一性较好,降低了对仪器系统的要求。 同时,固定的区间划分也为实时荧光检测提供了可能,可以提供更多、更准确的数据信息, 也具有更高的检测效率。但其缺点在于该系统对芯片制作的工艺要求较高,需要考虑芯片材 料对扩增效率的影响,提高了数字 PCR 分析的成本,且反应单元的数目受到芯片尺寸、制 作成本等因素限制,性能提升的空间有限。
“液-液相”数字 PCR 技术则是由二代测序中“油包水 PCR”技术发展起来的,其优势 在于液滴的体积可以从纳升级到皮升级进行变化,从而为“液-液相” 数字 PCR 系统提供 更高的反应通量和更好的扩展性,而且油包水的反应体系也具有更好的生物兼容性,能够提 供良好的 PCR 反应条件。但其缺点在于液滴的稳定性与可控性相对较差,液滴的少量融合 难以避免,对于检测分析过程也提出了更高的要求,现有仪器对液滴都采用终点检测,在一 定程度上也影响了数据的可信度。
1、基于集成流路(IFC)芯片的数字 PCR 仪器平台
通过高密度的微阀结构设计在芯片上制备出成百上千个皮升级的独立单元的芯片称为集成流路(integrated fluidic circuit.IFC)芯片。IFC 芯片发展背景微流控芯片技术是一种以微 米尺度空间对流体进行操控为主要特征的科学技术,具有将生物、化学等实验室的基本功能 微缩到一个几平方厘米芯片上的能力,因此又称为芯片实验室(lab-on-a-chip)。在现阶段,主 流形式的微流控芯片多由微通道形成网络,以可控流体贯穿整个系统,用以实现常规化学或 生物等实验室的各种功能。
基于聚二甲基硅氧烷( polydimethylsiloxane. PDMS) 微阀的 IFC 芯片具备高通量低消 耗、操作方便、准确率高等优点,为实现低成本、小体积和高通量平行 PCR 分析提供了一 个理想的平台。2006 年底,Fluidigm 公司率先推出了第一款具有数字 PCR 分析功能的产 品--基于 IFC 芯片技术的 Bio-MarkTM HD 系列高通量基因分析系统,并陆续开发出不同型 号的芯片平台,以满足基因表达图谱构建、拷贝数变异分析、稀有突变检测等不同应用需求。

图 7.2 Bio-MarkTM HD 基因分析系统
Bio-MarkTM HD 高通量基因分析系统是一款融合了 IFC 芯片技术、实时定量 PCR 技 术及强大的基因分析软件的仪器平台,同时具有实时荧光定量 PCR 分析和数字 PCR 分析 的功能,而其分析功能的选择则由其采用的 IFC 芯片类型决定。Bio-MarkTM HD 系统的核 心技术就在于将实时定量技术集成了流体通路技术:利用集成电路制作工艺(光刻)在硅片 或石英玻璃上刻上许多微管和微腔体做成阵列芯片,并通过独立的纳米级微型阀门控制溶液 在阵列反应仓(Reaction Chamber)中的流动来实现生物样品的分液、qPCR 体系混合建立, 并实现高效准确地实时 PCR 扩增、检测。IFC 芯片技术简化了生物样品和试剂的分液操作,能够有效提高分析通量和检测灵敏度。与此同时,其纳升级的反应体系大大降低了检测所需 的样本与试剂的消耗,在高通量基因分析中可以节省大量成本。
作为首款数字 PCR 仪器,Bio-MarkTM HD 高通量基因分析系统在检测的通量、灵敏度、 灵活性上取得了革命性的突破,为实时 PCR、终点 PCR 及数字 PCR 分析提供了极具兼 容性的新型检测平台。Bio-MarkTM HD 高通量基因分析系统由 4 部分构成: BiomarkTM HD 实时 PCR 仪(整合了高性能计算机)、IFC 芯片(耗材)、IFC 控制器(将生物样品与反应 试剂导人到 IFC 芯片中)和数据分析软件。其中,Bio-MarkTM HD 实时 PCR 仪采用 LED 激发光源和 CCD 光学检测器件,能够完成 4 个荧光通道(FAM、VIC 与 ROX 及选配的 Cy5)的实时荧光采集分析。针对不同应用,仪器系统也提供了强大的数据分析软件,可以 将数据以不同的形式展现出来,提供 IFC 芯片荧光信号的热点图或者芯片上每个反应单元 实时荧光扩增曲线,数据分析精简实用。Bio-MarkTM HD 系统能够兼容 2 种类型的 IFC 芯 片:Dynamic Array(48.48 动态阵列芯片、96.96 动态阵列芯片和 192.24 动态阵列芯片) 和 Digital Array(12.765 数字阵列芯片和 48.770 数字阵列芯片),以用于基因突变检测基 因分型、核酸定量、单细胞基因表达等不同的基因分析。其中,数字阵列 IFC 芯片用于实 现数字 PCR 检测功能,并在数字 PCR 分析过程中起到样品划分的作用。
芯片使用常规 PCR 方法与试剂,具有良好的兼容性与灵活性,在一张芯片上可同时分 析 12 个(12.765 数字阵列芯片)或 48 个(48.770 数字阵列芯片)样本,并将 PCR 试剂划 分成数百个独立的纳升级反应单元,开展数字 PCR 分析,提高了检测的灵敏度和准确性。 整个实验过程自动化程度很高,可以分为以下 5 个步骤:1准备芯片与待检样品;2将制备 好的待检样品移液至 IFC 芯片的进液口;3将芯片放人 IFC 控制器运行,通过仪器的软件 界面进行操作完成样品到各反应腔体的上样;4将芯片放人 Bio-MarkTM HD 实时荧光定量 PCR 仪,进行 PCR 扩增和实时荧光检测;5利用强大的分析软件包分析实验数据,获取 荧光热点图或各反应单元的扩增曲线。
与此同时,除了将实时荧光监测功能与数字 PCR 分析功能进行集成的 Bio-MarkTM HD 高通量基因分析系统,Fluidigm 公司还为用户提供了一整套基于 IFC 芯片进行终点法荧光 分析的数字 PCR 仪器系统-- EP1TM 分析系统。EP1TM 系统跟 Bio-MarkTM HD 系统类似,包 括 IFC控制器(将生物样品与反应试剂导入到IFC 芯片中)、热循环仪(PCR扩增)和 EP1 阅读仪(终点法荧光检测与分析)及 IFC 芯片等耗材,且只需要增加 IFC 控制器与执循环 仪的数量就可以在 1 台终占阅读仪上成倍地提高系统的检测通量,具有良好的经济性与灵活 性。在操作流程上,EP1TM 分析系统与 Bio-MarkTM HD 高通量基因分析系统没有太大区别, 只是将扩增与检测过程分离到 2 台仪器中完成,在扩增结束后进行终点法的荧光检测与分析。
无论是 Bio-MarkTM HD 高通量基因分析系统还是 EP1TM 分析系统,它们实现数字 PCR 分析的基础都在于通过数字阵列 IFC 芯片完成样品的纳升级划分操作,然后进步完成 PCR 扩增和荧光检测,这两种仪器系统都很好地实现了数字 PCR 分析的功能,唯一的区别在于 Bio-MarkTM HD 高通量基因分析系统在 IFC 芯片扩增的同时可以进行实时荧光监测,从而 能够获取更多的检测信息;而 EP1TM 分析系统只是对扩增后的 IFC 芯片进行终点荧光检测 与分析,但将扩增与检测过程分离也使得系统具有更好的灵活性。
相比早期的数字 PCR 系统,Bio-MarkTM HD 高通量基因分析系统和 EP1 分析系统借 助 IFC 芯片真正意义上实现了精细化的样品划分能够对极微量的试验对象同时进行成千上 万次复杂的生化测定,为研究人员提供高通量和高灵敏(即浓度低至单分子水平)的研究平 台,这对推动数字 PCR 技术的产业化应用具有很大的意义,也为后续的数字 PCR 仪器系 统设计提供了参考。
Fluidigm 公司的Bio-MarkTMHD系统和EP1TM系统通过配合IFC数字阵列芯片成为首 款实现数字 PCR 分析功能的商业化仪器平台,在单个芯片上能够同时分析 12 个或 48 个样 本,有着较强的荧光分析能力。同时,Bio-MarkTMHD 系统还具备实时荧光监测能力,可以 为用户提供实时荧光定量 PCR 分析和实时荧光监测的数字 PCR 分析的功能。但是,这两款 仪器系统价格较为昂贵,芯片耗材的价格也领一般实验室难以负担,而且目前相比较而言, 其数字 PCR 分析能力则显得较为薄弱,存在死体积较大,反应单元数较少,检测的灵敏度 和动态范围不足等问题。
2、基于微孔芯片的数字 PCR 系统
1)微孔芯片的技术背景
传统的 96/384 孔板无法满足数字 PCR 对样品划分精细化程度和数据群体体量的要 求,日在通量的提升上受到试剂成本和工作量成比例增加的限制。微纳制造工艺的发展使得 人们认识到可以通过制备高密度纳升级反应阵列来进行高通量 PCR 反应,而且能够同时兼 具高精度、高准确度和宽动态范围及多基因与样本并行定量分析的能力。基于在平面基材上 加工高密度微孔阵列的小型微孔板是实现纳升或皮升级 PCR 反应的手段之一。除此之外, 还可以通过材料表面的图案化的亲疏水修饰或二维的交叉微通道控制而形成微滴结构以进 行 PCR 反应。虽然这些方式为构建高密度且相互独立的反应单元(>4 个/mm),需要隔绝反 应单元间的流体流动并严格控制环境以防止温度循环过程中的交叉污染和蒸发损失,但是微 纳芯片的平行结构为 PCR 反应带来了通量优势,可以同时完成许多样本的热循环与荧光成 像,从而对靶标基因进行拷贝数定量。与此同时,并行 PCR 反应的成像检测过程可以有更 长的曝光时间,这将有助于改善检测的信噪比,并在更少的温度循环次数下提高检测给定靶 标拷贝数的 PCR 特异性和灵敏度。此外,当反应体积降低时,成比例增加的表而积体积比 将有利于热量的快速传递,大大缩短热循环的时间。
但要实现这上述目标,需要克服两大难题:一是如何实现微升级样品到纳升级反应单元 的精准划分:二是在反应体系降低到纳升级别之后如何保证检测的精度、准确度及灵敏度。 因此,找到一种能够实现反应试剂快速准确划分并保证纳升级尺度下 PCR 分析性能的方法 是当前的首要工作目标。
为解决这些问题,Morrison 等采用光刻与湿法刻蚀的加工工艺,在载玻片大小 (25mmx75mmx0.3mm)的不锈钢材料(317 不锈钢)上加工出 3072 个直径为 320μm 的 通孔(through-holes)结构,将每个微孔反应单元的体积降低至 33nL。这些通孔结构由 48 个 64 孔的子阵列组成,与 384 孔板的孔距相同,每个子阵列间距也为 4.5mm。经过一系列 的亲疏水处理方法,该微孔板的通孔内表面变得亲水而孔外疏水,亲疏水涂层的差异将促进 样品的精确加载与划分,并使得样品保留在微孔内而不会残留在芯片表面。通过后期的芯片 封装,就可以将该芯片用于 PCR 扩增与实时荧光检测。经实验证明,该微孔芯片与 384 孔 板的检测灵敏度相当,同时反应体积降低为原来的 1/64,分析的通量提高了 24 倍,且工作 流程能够与标准的微孔板兼容。
2)基于 OpenArray 微孔板的数字 PCR 分析平台
2010 年,Life Technologies 公司(现已被 Thermo Fisher 公司收购)对前述微孔阵列芯 片进行了产品化,并基于 OpenArray®实时荧光定量 PCR 系统推出了一款初具雏形的数字 PCR 分析平台。其中,OpenArray 微孔板是该系统的关键技术之一。OpenArray 微孔板由 48 个 64 孔的子阵列组成,包含 3072 个通孔结构,每个通孔都能作为独立的 PCR 反应单元。 单个 OpenArray 微孔板可分析 1-48 个样本,每个样本可以获取至少 64 个数据点。微孔板 经过表面亲疏水处理,使通孔外部疏水而内部亲水且具有良好的生物兼容性,从而在表面张 力的作用下促进样品的精确加载,保证 3072 个通孔中都分别包含 33nl 的 PCR 反应试剂。
目前,该微孔板可以在已有的 OpenArray®系统和 QuantStudioTM 12K Flex 系统上运行。 其中,具有 OpenArray®模块的 QuantStudioTM 12K Flex 系统主要由 QuantStudioTM 12K Flex 实时荧光定量 PCR 仪,QuantStudioTM 12K Flex OpenArray® AccuFillTM 系统套的计算机 系统与分析软件等组成。

图 7.3 QuantStudioTM 12K Flex 实时荧光定量 PCR 仪

图 7.4 QuantStudioTM 12K Flex OpenArray® AccuFillTM 系统
虽然 QuantStudioTM 12K Flex 本质上是一款实时荧光定量 PCR 仪器,但通过配合 OpenArray 微孔板使用,可同时运行 4 块 OpenArray 微孔板并产生 12 228 个 33nl 的独立 PCR 反应,还可以实时采集 PCR 扩增的荧光数据,相当于同时进行 32 个传统的 384 孔 板的 PCR 实验。QuantStudioTM 12K Flex OpenArray® AccuFillTM 系统于完成 PCR 试剂 由 384 孔板到 OpenArray 微孔板的自动化转移。此外,Life Technologies 公司还提供了用于数字 PCR 分析的 TaqMan® OpenArray® 试剂盒。
基于 OpenArray 微孔板在 QuantStudioTM 12K Flex 系统上进行数字 PCR 分析,其实 验过程如下。
(1)样本准备,分离纯化基因组 DNA。
(2)利用 OpenArray® 数字 PCR 软件协助完成实验设计,确定样品稀释程度,以获得 最优化的数字 PCR 定量结果。
(3)样本加载,将 DNA 样品与 TaqMan Assay 及 OpenArray® 数字 PCR 预混液加载 到 OpenArray 384 孔板,然后利用 OpenArray® AccuFillmTM 系统将反应试剂上样到 OpenArray 微孔板。
(4)热循环、荧光成像,将 OpenArray 微孔板封装到铝盒中,装满浸液并用胶水密封, 然后放置到QuantStudioTM 12K Flex 实时定量 PCR 仪中执行 PCR 反应。
(5 )采用数字 PCR 软件分析模块对数据进行分析,显示数字化的分析结果。
对于既开展实时定量 PCR 研究又需要开展数字 PCR 研究的实验室来说 QuantStudioTM 12K Flex 系统是个非常理想的选择。它可以根据实验需要选择并加载热循环 模块(支持标准或快速 96 孔、384 孔、TaqMan® Array Card 和 OpenArray® 模块)、无须 工具,不到 1 分钟即可完成模块更换,具有极大的灵活性。与此同时,QuantStudioTM 12K Flex 系统还采用增强型 OptiFlex® 系统(白光 LED 和 21 种滤光片组合),可实现可靠的荧光检 测,获得准确和灵敏的数据。QuantStudioTM 12K Flex 系统还为基因表达、基因分型和数 字 PCR 等应用提供了一整套软件分析工具。其中,针对数字 PCR 分析提供的 DigitalSuiteTM 软件采用泊松分布统计模型对实时荧光 PCR 的数据进行分析,获取每个通 孔的疏水 DNA 分子拷贝数并展示数字化的分析结果,实现了定量 PCR 到数字 PCR 的无 缝转化提高实验的精确度和灵敏度。
3)QuantStudioTM 3D 数字 PCR 仪器系统
虽然我们将基于 OpenAray 微孔板进行 PCR 分析的仪器称之为数字 PCR 仪器系统, 但其单芯片上 48 个 64 孔的子阵列(3072 个通孔)所构成的独立反应单元对于数字 PCR 高 精度、高灵敏分析尚显不足。因此,2013 年下半年,Life-technologies 在 QuantStudioTM 12K Flex 仪器系统之后推出全新的芯片式数字 PCR 系统-QuantStudioTM 3D 仪器系统。该仪器系 统在提高芯片与仪器性能的同时充分考虑了系统的稳定性与运行成本,并对仪器操作过程进 行优化,尽可能缩短手动操作的时间,促进数字 PCR 技术的普及应用。
相比其他的数字 PCR 仪器系统,QuantStudioTM 3D 数字 PCR 系统采用高密度纳升 级微孔阵列芯片技术和简单可靠的仪器设计,实现了样本的均匀分配和反应孔间的有效隔离, 具有良好一致性的微孔反应单元保证了实验结果的稳定性和可靠性,能够满足稀有突变检测、 基因组靶标间低倍差异分析或 GMO 检测与污染评估等绝大多数数字 PCR 应用分析的需 求。该仪器系统具有简化的工作流程及样本处理步骤,1 分钟上样、1 分钟读取分析;20 000 个反应单元可提供高精度和高灵敏的绝对定量分析结果;采用密封芯片和非暴露的样本转移 步骤,减少样本损失并有效降低样本污染的风险;并且其有良好的兼容性,使用现有的 TaqMan® 实时 PCR 分析试剂即可开展数字 PCR 实验,支持染料法检测;QuantStudioTM 3D 阅读仪可以直接对数据进行分析并在触摸屏上显示初步分析结果。

图 7.5 QuantStudioTM 3D 芯片阅读仪
QuantStudioTM 3D 数字 PCR 仪器系统由数字 PCR 芯片和包括芯片加样仪、PCR 热循 环仪、芯片阅读仪在内的 3 台仪器组成。其中,数字 PCR 芯片包含了 20000 个通孔微结 构,每个通孔都能作为独立的 PCR 反应单元;芯片加样仪可以自动化地实现试剂到 20000 个独立反应孔的均匀分配;ProFlex®PCR 热循环仪包含两块平板热学模块,最多可同时运行 24 片数字 PCR 芯片: QuantStudioTM 3D 芯片阅读仪能够在 1 分钟内完成对单个芯片上 20000 个独立反应单元的荧光信号采集与分析。
QuantStudioTM3D 仪器系统降低了数字 PCR 检测对用户专业水平的要求,通过样品制 备→芯片自动上样与密封→热循环扩增→芯片读取与分析 4 步简单的流水线操作方式,最大 限度地减少了移液操作,可以实现全程封闭操作并得到简单明了的结果(拷贝/微升)。其具体 操作流程如下:1通过混合样本、预混液、TaqMan®Assays 等制备 PCR 反应试剂;2将制备 好的试剂通过手动或自动加样仪加载到数字 PCR 芯片上,然后加盖并注入浸液油(immersion fluid)再通过紫外固化胶密封填充口;3采用双平板加热模块对芯片执行 PCR 热循环;4采 用 QuantStudioTM 3D 阅读仪读取并分析数字 PCR 芯片的荧光信号,并在仪器显示屏上显示 初始分析的结果(目标 DNA 分子的拷贝浓度);5根据需要,可以采用 QuantStudioTM3D AnalysisSuiteTM 分析软件对 QuantStudioTM 芯片综合分析。
QuantStudioTM 3D 芯片阅读仪为用户提供了简单直观的触屏操作界面,采用 CMOS 检 测器直接对数字 PCR 芯片进行二维光学成像,能够在 30 秒内完成对单个芯片的三通道荧 光图像采集,然后需要下一个 30 秒时间完成数据的初步分析,并在触摸屏上品示样本的浓 度(拷贝/微升)。QuantStudioTM3D 阅读仪能够并行完成荧光成像和数据分析的功能,即在 分析前一个数字 PCR 芯片的荧光图像数据的同时依然可以进行下一个芯片的荧光采集。在 分析完成后,阅读仪将提供芯片 FAM/VIC 荧光通道所对应的核酸靶标浓度评估结果,并 通过颜色编码(绿、黄、红)的旗形标志显示初始分析过程中数据质量的简单评价。
对于每一个芯片,阅读仪都将获取的荧光图像数据和初始分析结果生成并保存成一个与 芯片编号对应的实验文件(.eds),并根据数据终端设定选择数据文件保存的位置,或自动 地上传到个人 QuantStudioTM3D AnalysisSuiteTM 的云端账户,或传输到网络文件服务器,或 保存在 U 盘中,以进行后续的 AnalysisSuiteTM 软件分析。
AnalysisSuiteTM 软件包含了多个分析模块,用户可以采用绝对定量模块进行诸如分子标 准品定量、病原体与病毒载量分析等研究,提供样本的拷贝浓度检测结果和上、下置信区间 及精度统计数据,还可以采用相对定量模块进行基因组靶点间的低倍差异分析拷贝数变异分 析。此外,AnalysisSuiteTM 软件能够将高达 100 个数字 PCR 芯片的数据合并分析用户既 可以结合质量控制特性对单个芯片进行数据审查,也可以通过相对定量模块将多个芯片的数 据依据 FAMVIC 荧光信号自动地合并为一张散点图,然后手动定义 FAM/VIC 及不确定荧 光信号的范围(阈值),并利用软件对所有芯片进行突变型和野生型基因比率分析。
QuantStudioTM3D 数字 PCR 系统为用户提供了性能稳定且具有较高性价比和较低运行 成本的仪器系统,对推动数字 PCR 的普及应用有着积极意义。与此同时,该系统采用高密 度固态纳升微孔芯片,保证样品划分的一致性和可靠性,为提供高精度、高灵敏度数字 PCR 分析及可靠的实验结果奠定了扎实基础。目前,QuantStudioTM 3D 数字 PCR 系统已经在循 环肿瘤 DNA 检测、稀有突变检测和拷贝数变异分析等需要进行核酸定量的研究分析中得 到广泛应用。
3、液滴数字 PCR 系统
1)QX100 TM /QX200 TM 微滴式数字 PCR 系统
QuantaLife 公司首先利用油包水微滴生成技术开发了微滴式数字 PCR(droplet digital PCR,ddPCR)系统,并因此获得了 2011 年度 Frost&Sullivan 北美新产品创新奖。2011 年 10 月,Bio-Rad 公司收购了 QuantaLife 公司和 ddPCR 技术,并迅速推出 QX100 TM 微滴式 数字 PCR 系统(QX100TM droplet digital TM PCR)。QX100TM 系统采用独特的微滴化技术 能够将单个样本分成 20 000 个高度均一的纳升级液滴单元,本质上将传统定量 PCR 的单 个反应变成 20 000 个反应,大大提高了痕量核酸序列检测的灵敏度。QX100TM 系统为数字 PCR 概念的普及和应用领域的拓展发挥了重要作用,在稀有突变序列检测、病原微生物检 测和拷贝数变异遗传分析、转基因食品检测等方面有着诸多应用实例与文献,显示了“第三 代 PCR”技术广泛的应用范围与强大的生命力。在全球竞争最激烈、最成熟的北美市场 QX100TM 系统的市场占有率遥遥领先,并因此获得 2012 年度 Frost & Sullivan 北美地区数字 PCR 市场领导者奖项。
在此基础上,Bio-Rad 于 2013 年 8 月 14 日在全球正式推出其第二代的液滴数字 PCR 系统--QX200TM 微滴式数字 PCR 系统。该系统除秉承 QX100 TM 系统的优良特性之外,还 增加了对 EvaGreen 染料法的支持,使之可以更好地与定量 PCR 方法兼容,有效降低实验 成本。此外,QX200TM 系统采用硅光子计数器(MPCC)代替 QX100TM 系统中配置的光电 倍增管(PMT),使其具有更优秀的单光子分辨能力。QX200TM 微滴式数字 PCR 系统可对靶 标 DNA 或 RNA 分子进行绝对定量分析,适用于基于 TaqMan 探针或 EvaGreen 染料的 数字 PCR 应用领域,能够实现超高灵敏度或高通量的样品分析。

图 7.6 QX200 TM 微滴式数字 PCR 分析仪及微滴生成器
QX200 TM 微滴式数字 PCR 系统的优点:1准确、灵敏的数字 PCR 解决方案,满足不 同的应用需求;2灵活的数字 PCR 化学方案,已针对 TaqMan 水解探针和 EvaGreen 测 定进行优化;3灵活的实验设置,可通过扩展实现较高的灵敏度或通量;4简便易用的实验 流程,单次运行通量可达 96 个样本;5QX200TM 微滴数字化技术可减少扩增效率和 PCR 抑制物带来的偏差;6实验设计方便,不需要标准曲线。
QX100TM/QX200TM 微滴式数字 PCR 系统结合了油包水乳化微滴技术与微流体技术, 其系统主要由微滴生成器(droplet generator)、微滴分析仪(droplet reader)和热循环仪及相 关的试剂与耗材组成。微滴发生器通过一次性耗材--微滴发生卡可最多同时处理 8 个样品, 并将每个样品划分成 20 000 个均一的纳升级液滴,其中每个微滴或不含待检核酸靶标分子, 或者含有 1 个至数个待检核酸靶分子,且每个微滴都作为一个独立的 PCR 反应单元。随后 包含微滴的乳液被转移到 96 孔 PCR 板中,在常规 PCR 热循环仪上完成扩增。PCR 扩增后, 微滴分析仪对微滴逐个进行荧光检测,识别出阴/阳性的微滴,最后根据泊松分布统计原理 及阳性微滴的比例计算出待检靶标分子的拷贝浓度。目前,QX100TM/QX200TM 微滴式数字 PCR 系统的应用涉及了肿瘤机制研究个体化医疗及疾病的超早期分子检测、拷贝数变异遗 传分析和基因表达分析等多个研究领域,为遗传病、癌症、传染病的研究提供了一种全新的 技术思路与手段。
在已推出的两代微滴式数字 PCR 系统的基础上,Bio-Rad 公司还从多个角度对其微滴式 数字 PCR 系统进行提升,以满足各个领域用户的数字 PCR 应用需求。一方面,Bio-Rad 公 司经过严格的实验室验证,按照 MIQE 原则专门为微滴式数字 PCR 系统设计开发了即用型的 PrimePCR TM ddPCR 系列试剂盒,研究人员可以根据自己的研究需求(包括突变检测和拷 贝数变异分析)将仪器系统与这些经过验证的试剂盒结合起来。另一方面,为了更好地简化 微滴式数字 PCR 的操作流程,Bio-Rad 公司在 OX200 TM 微滴式数字 PCR 系统的基础上推 出了对应的全自动型号(QX200 TM AutoDG TM Droplet Digital TM PCR 系统)。该系统利用自动 化的微滴生成器(automated droplet tenerator,间称 AutoDG)替代原有的微滴生成器,并配合 QX200m 微滴分析仪完成液滴数字 PCR 分析功能。
该全自动型号的面市大大简化微滴生成的步骤,为 QX2000 TM 微滴式数字 PCR 系统提 供高通量的微滴制备方案,在 45 分钟内即可全自动完成 96 个样品的微滴生成,通过一个 彩色的触摸屏进行程序设置与仪器控制,操作简便;在微滴生成的过程中,能够消除不同操 作者引入的人为操作误差,确保微滴发生的一致性;此外,AutoDG 仪器有其自己的罩子和 空气过滤器(high efficiency particulate air filter,HEPA),为微滴制备提供清洁空间,降低了 交叉污染的风险,也降低了对实验室环境的要求。
QX100 TM /QX200 TM 微滴式数字 PCR 系统基于微液滴技术和微流体技术来实现数字 PCR 分析,整个分析过程除少量试剂转移操作外完全由仪器自动化地完成微滴的制备、油 包水乳液的 PCR 扩增、液滴流式荧光检测和数据分析。微滴式数字 PCR 系统的工作流程 如下:
(a)制备 ddPCR 反应液,将核酸样品、引物、探针与 Bio-Rad 的 ddPCR TM 预混液均 匀混合,在微滴发生卡的各组样品孔和油孔中分别加入 20μl 反应液和 70μl 微滴生成油。
(b)制备液滴,将微滴发生卡放入微滴生成器,单次运行可同时处理 8 个样品,并在 2.5 分 钟内制备出 8x20 000 个微滴,核酸序列随机地分布到各个微滴内。
(c)PCR 扩增,将经过微滴化处理的样品转移到 96 孔 PCR 反应板中,封膜,然后采 用常规 PCR 热循环仪执行 PCR 扩增(约 40 个循环)。
(d)液滴荧光检测,将扩增后的 96 孔板放入微滴分析仪,仪器将通过钢针按顺序吸取每 个样品的微滴,并采用流式检测技术使微滴随油相鞘液逐个通过双色荧光检测器,读取每个 微滴在 FAM(EvaGreen)与 HEX(VIC)荧光通道下的荧光强度(每次运行检侧 96 个样本)。
(e)荧光数据分析,系统软件自动完成对微滴的阴/阳性判别并对 PCR 阳性微滴和 PCR 阴性微滴的数量进行计数,然后根据阳性微滴的比例并结合泊松分布统计原理计算出靶标 DNA 的绝对拷贝浓度。此外,软件还可以对多个样品的平均值和总误差进行统计分析,或 者合并多个样品的微滴总数进行统计分析。
(f)结果显示,系统软件可以以多种方式显示检测结果,例如给出每个样品中含有靶标 DNA 分子的起始拷贝数或浓度(拷贝/微升)或者显示每个微滴在 FAM 通道和 VIC 通道的 荧光强度分布等,检测结果也可以直接打印输出或导入 Excel 表格。
Bio-Rad 微滴式数字 PCR 系统采用功能强大的 QuantasoftTM 分析软件配合微滴分析 仪对每个微滴进行荧光检测和数据采集与初步分析。QuantasoftTM 分析软件提供了友好的操 作界面,用户可以根据实验分析的要求对各个反应孔的样本、实验类型(包括绝对定量、稀 有突变检测、拷贝数变异和基因表达分析)及荧光通道进行设置定义。随后,软件将按顺序 对各个样品的数据进行初步分析,在分析过程中实时显示 2 个荧光通道的信号采集情况(无 论是单通道还是双通道实验均会同时显示散点图和频率分布图),并显示初步计算的核酸浓 度结果,全部运行完成后则会对数据重新进行分析。
随后,用户可以导入并分析之前存储的数据,也可以直接对运行完毕的结果进行分析, 在软件界面上显示选定样品孔的各种图形化数据结果,如各样品的一维通道数据、二维聚类 散点图(clustering plots)、95% 置信水平下的浓度值与误差线(error bars )、拷贝数分析结果及 阴阳性微滴个数统计等。
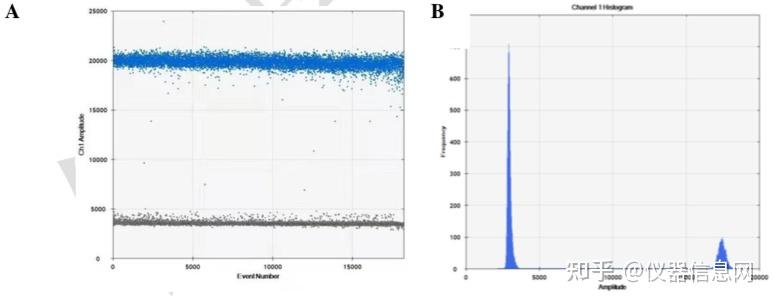
图 7.7 荧光通道的一维数据图 A.荧光散点图 B. 微滴频率分布图
图 7 中,左侧的荧光散点图的横坐标代表微滴数,纵坐标代表荧光强度,阴性微滴以灰 色显示,阳性微滴以颜色显示(默认 Ch1 蓝色,Ch2 绿色),在热点图模式下则根据微滴密 度显示不同颜色;右侧的微滴频率分布图,横坐标代表荧光强度,纵坐标代表微滴数。图中 阴/阳性微滴的判定是由荧光阈值大小决定的,软件会自动计算(auto analyze)网值并判定微滴的阴/阳性,但在面临特殊实验情况时,自动的域值设定会存在较大计算误差或无法进行自 动化的阈值设定,这时就需要用户针对单个或多个样品手动设定國值。
数字 PCR 因其良好的精确性与可重复性及在诊断和药物研究相关应用上的潜力而引 起了行业的极大关注,而 Bio-Rad 公司推出的液滴式数字 PCR 仪器系统则以其独特的优 势,获得了较大的市场份额,在数字 PCR 技术的推广与普及应用上发挥了非常重要的作用。 该系统所采用的微滴技术让数字 PCR 成本更低且更加实用,克服了数字 PCR 系统检测成 本高、通量有限且流程复杂等缺陷。其优势在于能够重复产生非常均一的纳升级液滴,将每 个样品划分成 20 000 个微滴单元,并保证其在热循环扩增过程中的稳定性。这样就比大多 数的数字 PCR 仪器系统具有更多的反应单元,也意味着分析更加准确,定量精度更高。目 前,Bio-Rad 公司的 QX100TM/QX200TM 微滴式数字 PCR 系统在拷贝数变异研究稀有突变 序列检测和血浆游离 DNA 检测与定量等应用领域的作用已经得到充分验证。同时,微滴 式数字 PCR 系统还能够与二代测序/全基因组测序技术进行整合,与测序结果进行相互验 证,并且可以进行 NGS 文库的制备与定量,充分展现了该系统的良好平台基础和优异分析 性能。除此之外,Bio-Rad 公司也不断对其微滴式数字 PCR 系统进行软硬件的升级,并针 对不同应用研究开发出相应的试剂盒,为研究人员提供一整套简便易用、高准确度、高灵敏 度的数字 PCR 解决方案。
Bio-Rad 依次推出的 QX100TM 系统、QX200TM 系统及 QX200TM Auto DGTM 系统, 在不断提高仪器自动化操作水平的同时,也改善了仪器系统的试剂兼容能力。这几款系 统基于微液滴技术实现数字 PCR 分析,提供了稳定的纳升级微滴反应系统,仪器成本和 运行成本较低(无芯片、无激光),并且具有较高的运行通量和良好的数字 PCR 分析能 力,也是目前市场占有率最高的数字 PCR 系统。不过 QX100TM/QX200TM 系统在运行过 程中需多次移液操作,这对于微滴反应体系来讲容易产生微滴融合、损失等一系列不良 影响,并可能进一步影响检测结果的精确度。虽然后来出现的 QX200TM Auto DGTM 系统 已经实现液滴制备与转移过程的自动化,但仪器系统价格也有较大幅度的提升,不利于 系统的推广与普及。与此同时,目前 Bio-Rad 的液滴数字 PCR 系统必须使用 Bio-Rad 提 供的专用试剂,也限制了系统在特定场合的使用。
2)RaindropTM 微滴式数字 PCR 系统
RainDance 公司成立于 2004 年,拥有基于油包水技术的 100 多项发明专利。2012 年 4 月,RainDance 公司在美国癌症研究学会(AACR) 的年会上推出了高灵敏的 RaindropTM 数字 PCR 仪器系统,如图 8 所示。RaindropTM 微滴式数字 PCR 仪器系统由微滴制备(source) 仪和微滴分析(sense)仪及配套 PC 工作站和操作软件组成,并配备微滴制备芯片和微滴分析 芯片及配套试剂,包含微滴的乳液则采用常规 PCR 热循环仪进行扩增。其中,微滴制备仪 配合微滴制备芯片使用,包含 8 孔样品输入、输出位置,可以在每小时内处理 16 个 25μl 体 系的样品,仪器内置照相装置和图像分析软件,以每秒 15 帧的捕捉速率,实时分析监控微 滴生成和处理过程,确保微滴质量;微滴分析仪采用正压系统控制进样,配合微滴分析芯片 使用,同样包含 8 孔样品输入、输出位置,在 1000 000 个微滴/样本的检测模式下,每天可 检测 24 个样本。

图 7.8 RaindropTM 数字 PCR 仪器系统
类似于 Bio-Rad 公司的 QX100TM /QX200TM 数字 PCR 系统,RaindropTM 系统同样采用 油包永液滴生成和液滴流式荧光检测技术,将 50μl的 PCR 反应体系划分成 1000 万个皮 升级液滴,大大超过其他数字 PCR 系统。扩增完成后,微滴分析仪通过流式荧光检测技术 逐个检测液滴的荧光信号强度,统计阴/阳性液滴个数并结合泊松分布统计原理即可轻松计 算出样品的起始模板拷贝数,实现对模板 DNA 的绝对定量。
虽然同属于液滴数字 PCR 系统,但 RaindropTM系统同样有其特色与创新之处。首先, RainDance 公司拥有成熟的 RainStormTM微液滴技术,以该技术为基础的 RaindropTM微滴 制备仪能够快速高效均一地制备出高达 1000 万个皮升级大小的液滴,展现了其卓越的油包 水分配能力,大大提高了系统的精度、灵敏度和动态检测范围;其次,Raindrop TM 系统的微 滴制备与检测分别基于一次性芯片耗材--微滴制备芯片和微滴分析芯片来完成可同时处理 1-8 个样品,芯片集成有 12 个纳米级微流体滤器,可去除加样过程中混入的固体微粒,保证实验结果的可靠性;同时,Raindrop TM 系统提供了一个开放的数字 PCR 系统平台,能够兼容市场上大部分供应商提供的 PCR Master Mix 试剂;仪器系统采用闭管设计理念,微滴 制备仪制备的油包水微滴直接流入 PCR 管中,封好管口采用常规 PCR 仪进行闭管扩增, 扩增后的产物团管放人到微滴分析仪中进行检测,无须担心气溶胶污染,确保检测质量。此 外,RaindropTM系统利用 VIC 和 FAM 两种荧光标记并设置不同荧光探针浓度即可对同一 样本实现高达 10 重的 PCR 反应与检测,无须分管操作,避免分管检测造成的假阴性,同 时也降低耗材成本。该系统在数字 PCR 分析的灵敏度、绝对定量和多重分析方面具有优异 的表现,可应用于稀有突变检测、拷贝数突变检测、病毒含量测定、游离 DNA 检测及产 前检测等应用研究领域。
RaindropTM数字 PCR 系统具有较高的集成性和自动化水平,为用户提供了简便的数字 PCR 实验操作流程,研究人员只需要将配制好的 PCR 试剂转移到微滴制备芯片中,后续 实验过程中无须移液器操作,只需要将样品在不同仪器之间切换即可。Raindrop TM 数字 PCR 系统的实验操作流程,其详细过程如下。
(a)实验设计:根据分析需求,调整针对不同基因的荧光素浓度,轻松实现多重 PCR 检 测。
(b)配制 PCR 试剂:系统能够兼容市面上已有的大部分供应商提供的 PCR Master- Mix, 与 DNA 模板、引物和探针等均匀混合。
(c)微滴制备:将 PCR 试剂转移到微滴制备(source)芯片的样品孔内,source 芯片一次最 多可同时处理8个样品,然后将1个8 联排 PCR 空管和 source 芯片依次放入微滴制备仪, source 芯片的样品出口与 8 联排 PCR 空管对应。在高压气体的驱动下,PCR 试剂与包含 表面活性剂的氟碳油混合,制备出 5pl 大小的微滴,最后流人 8 联排 PCR 管进行收集。 对于 25μl 的 PCR 试剂,微滴制备时间需要 30 分钟左右,微滴制备过程可全系程实时分 析监控,以确保液滴单元的质量。
(d)热循环扩增:PCR 管采用硅胶盖密封并放人标准 PCR 仪进行扩增,包含模板 DNA 分子的微滴能够扩增从而发出荧光信号(即 PCR+微滴),不包含模板 DNA 分子的微滴则无 法扩增(即 PCR-微滴)。
(e)荧光检测与分析:将扩增后的 PCR 管和 sense 芯片依次放入微滴分析仪器 PCR 管 和 sense 芯片连接后进行流式荧光检测,微滴分析仪器可在 3.5 小时内逐个读取微滴在 FAM 和 VIC 通道下的荧光强度。数据结果以二维散点图表示,软件可以对每个基因理的 聚类液滴设门(gating),给出每个门(gate)中的液滴数值。
RainDropTM 数字 PCR 系统的微滴分析仪将检测到的数据结果以流式细胞术标准格式 ( fow cytometry standard,FCS)输出,能够兼容流式细胞仪分析软件。同时,Raindance 公司 也为该系统提供了配套的数据分析工具--RainDropTM Analyst II软件,以实现拷贝数变异、 基因分型、SNP 检测、病毒载量分析、核酸浓度的绝对定量等领域的分析功能。用户通过 该软件可以很方便地实现自定义的数据分析和数据输出设置,图形化的数据表示方法(如散 点图、三维图和自定义的报告)也可以让用户迅速直观地了解分析结果。这些图形化的分析 结果可以以 png、jpg、tif、emf、pdf、svg 等多种格式输出,也可以将分析结果以 Microsoft Excel、html、xml、text、csv、sql、xml 等格式的数据进行输出。
凭借其高灵敏度、多重分析能力、开放平台、闭管设计、简单操作等优势,RainDropTM 数字 PCR 系统能够应用于低频等位基因突变检测,miRNA 表达分析、DNA 甲基化检测 和 SNP 分组检测等研究领域,从根本上提升了现有分子检测平台的性能,受到国内外用户 的广泛关注。通过数百万个皮升级液滴反应的处理,RainDrop TM 数字 PCR 系统大大提高 了数字 PCR 分析的灵敏度。Pekin 等使用 RainDropTM 数字 PCR 系统对复杂背景下的稀有 突变进行检测,能够在高达 200 000 拷贝的野生型基因中检测到单拷贝的 KRAS 基因突变, 证明了 RainDropTM 数字 PCR 系统检测结直肠癌中 7 个 KRAS 突变位点,从患者血浆中得 到的循环 DNA,通过调节探针 FAM 和 VIC 浓度比例实现多重数字 PCR 分析。该研究 证明多重数字 PCR 可无创、高灵敏地检测循环肿瘤 DNA,也体现了 RainDropTM 数字 PCR 系统突出的多重分析与检测能力。在 CNV 的检测方面,Zhong Q 等同样利用 RainDrop TM 数字 PCR 系统对 20 例临床样品进行 5 重数字 PCR 检测,使用 FAM 和 VIC 两种荧 光分别测定 3 个基因(SMN1、 SMN2,BCKDHA)和单个核苷酸的突变(c.815>G、SMN1), 准确检测到基因缺失(SMN1),基因拷贝数变化(SMN2)及单个核苷酸的突变(c.815 >G. SMN), 充分验证 RainDropTM 系统通过多重数字 PCR 进行拷贝数变异分析和单碱基核薪酸突变检 测的功能。
4)锐讯生物 DropDx-2044 系统
2020 年 6 月,锐讯生物正式推出了 RainSure DropDx-2044 系统,该产品由 PCR 扩增 仪与生物芯片阅读仪两部分组成。PCR 扩增仪革命性地采用了一体式芯片设计,通过设备自动化完成样本的微滴生成与 PCR 扩增,整个实验过程,实验者仅需完成加样与芯片转移 操作。生物芯片阅读仪可自动检测、分析数据,具有四色荧光通道,能够检测更多试剂盒组 合,简化了多重数字 PCR 的设计难度。DropDx-2044 数字 PCR 系统采用全景式扫描模式, 无死角、无遗漏,迅速采集荧光信号,统计液滴数量,根据泊松分布自动计算核酸拷贝数浓 度,并自动处理成 CSV 数据报告。DropDx-2044 数字 PCR 系统获得了 NMPA、FDA-EUA 和 CE-IVD 三大注册证大满贯的数字 PCR 产品。
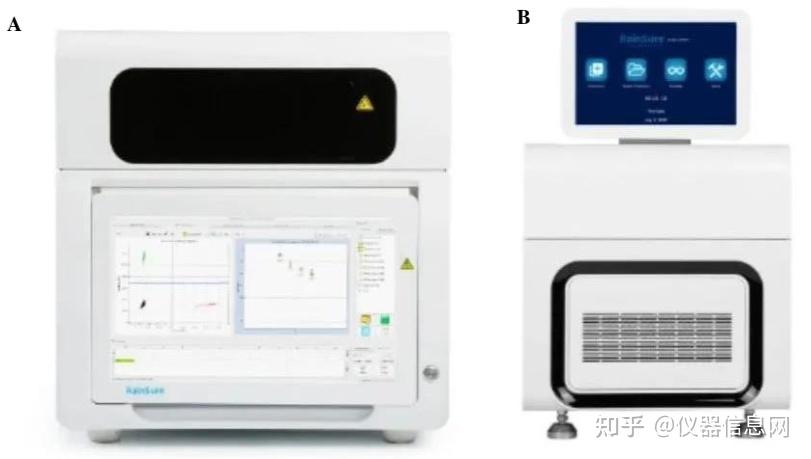
图 7.9 锐讯生物 DropDx-2044 系统 A.生物芯片阅读仪 Dscanner4-1000 B. PCR 扩增仪 SG-2000
PCR 扩增仪 SG-2000,运用自主研发的专利微流控乳液滴生成技术和独特的试剂体系, 快速实现油包水微乳液滴的制备。液滴的粒径均一、性能稳定,数目和尺寸可根据用户需求 灵活调整。整个制备过程全自动化,一键式操作,并且采用国际顶级温控原件,结合锐讯自 主研发的温控管理系统、一体式数字 PCR 芯片,实现对基因的快速扩增。
结合高精度压力控制微流控系统与微流控芯片,快速、平稳地产生粒径均一的“油包水” 液滴。可在 70s 内产生 20000-30000 个液滴,性能稳定持久:液滴尺寸均一,CV≤±3%,液滴 直径可根据用户需求在 70-110 微米范围内调整。采用全新的 PCR 芯片平板式加热设计,快 速升降温,最快升温速度达到了 10°C/s,最快降温速度达到了 6°C/s,45 分钟之内实现 40 个 PCR 热循环,比同类型品提速 2-3 倍 。
生物芯片阅读仪 Dscanner4-1000,具有四个荧光通道,运用精湛的光学技术设计,可以 准确快速地读取液滴的荧光信号值,提供阳性、阴性液滴数绝对数值。采用业界领先的四色 荧光通道设计,提升了数字 PCR 系统对多重 PCR 的包容性;多通道荧光信号采集设计,快 速、全覆盖;集成自动化图像分析处理系统。
4、NaicaTM 液滴数字 PCR 系统
1)NaicaTM 系统简介
Stilla Technologies 公司创建于 2013 年,基干在液滴微流控领域多年的研究和创新成 果,Stilla Technologies 公司致力于开发一款具有突破性意义的高分辨遗传分析工具 --NaicaTM 数字 PCR 系统。该系统首次采用液滴阵列(crystal digital PCR)的策略,将数字 PCR 分析过程集成到一个简单且高性价比的芯片耗材上实现,为用户提供了一个紧凑、简 单易用、快速和可靠的数字 PCR 的解决方案。

图 7.10 NaicaTM 数字 PCR 系统
如图 10 所示,NaicaTM 数字 PCR 系统由 Naica Geode 和 Naica Prism3 2 台仪器及 Sapphire 芯片组成。Sapphire 芯片在 Naica Geode 仪器中先后完成液滴阵列的制备和扩增. 然后转移到 Naica Prism3 仪器中进行荧光的采集以及后续的分析。通过构建液滴阵列的策 略,再结合强大的图像分析和直观的目视检测,该系统能够在数字 PCR 检测过程中获得较 好的置信度,提供值得信赖的数据分析结果。NaicaTM 系统具有以下三大优势:
(a)易用且集成的数字 PCR 解决方案:一次性的耗材;整个分析流程只需 2 台仪器
(b)快速便捷的数字 PCR 分析系统:包括热循环过程在内,数字 PCR 分析可以 2 小时内 完成并得到结果;手动操作时间低于 5 分钟。
(c)值得信赖的多重分析结果:三色荧光检测通道:友好的软件分析界面。
2)NaicaTM 系统运行流程
通过使用 Sapphire 芯片,NaicaTM 数字 PCR 仪器的运行变得十分的简单灵巧、快速有 效,用户只需要在实验开始前将 PCR 试剂加入到芯片的各个样品孔中并对芯片进行密封, 液滴生成与热循环扩增和检测分析将分别由 NaicaTM 仪器系统的 2 台仪器自动地完成,减少 了手动操作过程。与此同时,整个实验过程采用闭管操作的方式,有效避免了样品的污染, 提高了实验结果的置信水平。此外 Sapphire 芯片采用的阵列式液滴排布方式也有利于 CCD 荧光图像采集,能够为用户提供直观的荧光检测结果和可靠的荧光数据分析
该数字 PCR 仪器系统实验流程如下:
(a)实验准备,制备 PCR 反应混合液,并准备好芯片。
(b)将反应混合物加载到各个 Sapphire 芯片的样品孔中,密封后,将芯片放入 Naica Geode 仪器。
(c)启动仪器,制备液滴并随后执行 PCR 扩增。
(d)将扩增后的芯片转移到 Naica Prism3 液滴阵列阅读分析仪上,分别在三色荧光通道下读取液滴阵列的荧光信号。
(e)利用 Crystal MinerTM 软件进行目标核酸浓度的绝对定量。
其中,仪器 Naica Geode 和 Naica Prism3 的最高运行通量都为 12 个样本,液滴阵列 的制备需要 15 分钟,扩增过程需要 60 分钟,Sapphire 芯片上单个样本的荧光信号采集 与数据分析分别都需要 75 秒的时间,只需要 30 分钟即可完成 3 个芯片(12 个样本)的荧 光信号采集与数据分析。
4)NaicaTM 系统应用分析及评价
对于生物样本,常常会面临样本可用性十分有限的情况,因此对于研究人员和临床医生 来说,通过单次检测获取尽可能多的信息是非常必要的。针对这种情况,NaicaTM 数字 PCR 仪器系统提供了多重分析的能力,在影响结果的精度和可靠性的情况下通过单次测试即可对 多种生物标志物进行检测与定量。基于三色荧光检测通道,Naica TM 系统可以明确区分出不同的靶标分子,这一快速易用的解决方案能够兼容多种组合的荧光素,极大地扩展了实验设计的可能性。
在非小细胞肺癌中,表皮生长因子受体(EGFR)是一个重要的药物作用靶点。 EGFR 激 活突变,如 L858R 与 L861Q 突变是对采用酪氨酸激酶抑制药(TKIs)进行靶向治疗的疾病响 应前兆。相反,EGFR T790M 突变的出现与肿瘤对 TKIs 的耐药抗性相关。
为了通过一个简单的实验来监测患者对于 TKIs 治疗的反应,研究人员基于 NaicaTM 系统设计了一个能够同时分析 EGFR L858R、L861Q 和 T790M 及野生型 EGFR 靶标的多 重数字 PCR 实验,能够从野生型基因的背景中成功可靠地检出浓度低至 0.25 拷贝/微升(最 终浓度)基因突变,这意味着可以检出 0.05% 的等位基因突变。
虽然 Stilla Technologies 公司的 NaicaTM 仪器系统是一款极具创新意义的微额式数字 PCR 系统,其同样存在着先天的优势与不足。首先,由于仪器系统基于 Sapphire 芯片完成 液滴阵列制备、扩增和检测等一系列数字 PCR 分析过程,因此 NaicaTM 系统具有优良的集 成性能,且仪器操作简单快捷, 将整个数字 PCR 分析过程压缩至 2 小时内完成。与此同时, 液滴的制备、扩增和荧光检测都在芯片上进行也提供了全封闭的实验过程,避免了手动转移 操作带来的各种问题。此外,采用 CCD 图像传感器完成对芯片上阵列液滴的荧光采集, 能够为用户提供直观且可视化的荧光图像,这不仅提高了荧光检测的速度,也为后续的荧光 数据分析打下良好基础,使得实验结果更加精准可靠。但 Sapphire 芯片集液滴阵列制备、 扩增和检测功能于一体的设计理念虽然为仪器系统带来了诸多优势,但也从根本上限制了系 统的运行通量。
5、数字 PCR 仪器比较
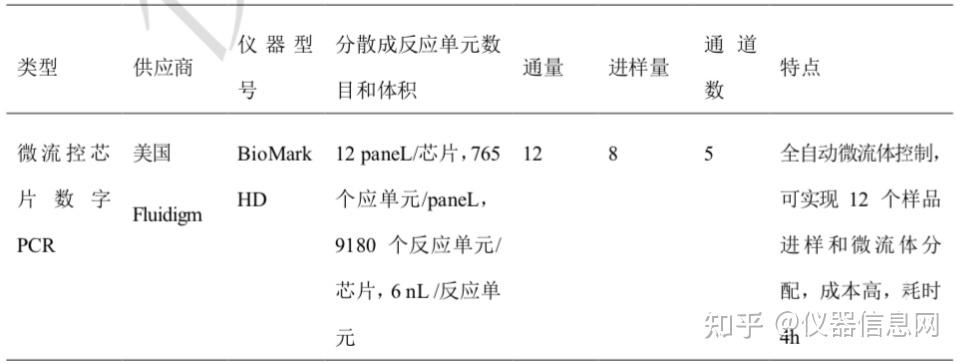


表 7.3 数字 PCR 仪器比较
第八章 原位PCR
第 1 节 原位PCR
一、简介
原位 PCR 技术起源于传统 PCR 技术和原位杂交技术。1969 年 Gall 和 Pardue 建立了核酸分 子的原位杂交技术(ISH)。该技术能对细胞或组织内的核酸或病毒进行定位和检测,具有 较高的灵敏度和准确的定位能力。聚合酶链反应(Polymerase Chain Reaction,PCR)是 1985 年出现的一项体外扩增核酸片段的基因检测技术。原位 PCR 是 Hasse 等人于 1990 年建立的 在组织细胞里进行 PCR 反应的技术,它是把 PCR 的高效扩增技术和原位杂交的细胞定位技 术相结合起来的原位多聚酶链式反应技术(in situ PCR),简称原位 PCR。
原位 PCR 结合了具有细胞定位能力的原位杂交和高度特异灵敏的 PCR 技术的优点。既能分 辨鉴定带有靶序列的细胞,又能标出靶序列在细胞内的位置,在分子细胞水平和分子水平的 研究上具有重大的实用价值,在众多学科领域中具有广泛应用。
二、起源
原位杂交技术是 Gall 和 Pardue 于 1969 年建立的一项把分子生物学、组织化学及细胞学相结 合的检测技术。原位杂交技术具有良好的组织定位能力,可以揭示出扩增的目的片段与目的 组织之间的关系,但其敏感性较低,对低浓度的目的基因检测能力较差。Mullis 于 1983 年 首先提出关于 DNA 扩增的设想,并于 1985 年发明了聚合酶链反应,即简易 DNA 扩增法, 意味着 PCR 技术的真正诞生。1986 年 Mullis 等将传统 PCR 技术进行优化完善,发现其能 够将微量的 DNA 或 RNA 进行大量扩增并且具有高度的特异性和敏感性。但传统 PCR 技术 需要将组织或细胞进行破碎,从中提取出核酸为模版进行扩增,不能在细胞内进行精准定位 且不能反映出扩增目的产物与组织之间的关系。
1990 年 Hasse 等首次通过将 PCR 技术和原位杂交技术结合起来检测了羊绒毛膜络从细胞中 的绵羊脱髓鞘脑炎病毒,建立了原位 PCR 方法。原位 PCR 既可以对含有目的片段的组织或 细胞进行精确的定位从而揭示出目的片段与组织或细胞间的形态结构信息,又由于其具有高 度的敏感性可以检测出组织或细胞中低浓度的 DNA 或 RNA,成为了细胞学科研与临床诊断 领域里的一项有较大潜力的新技术。
三、原理
传统 PCR 技术是一种在体外模拟体内 DNA 复制的核酸扩增技术,由于其高度的特异性和 敏感性, 能够将微量的 DNA 或 RNA 进行大量扩增。它以少量的 DNA 分子为模板,经过变 性-退火-延伸的多次循环,以接近指数扩增的形式产生大量的目标 DNA 分子。原位杂交技 术的基本原理是利用核酸分子单链之间有互补的碱基序列,将有放射性或非放射性的外源核 酸(即探针)与组织、细胞或染色体上待测 DNA 或 RNA 互补配对,结合成专一的核酸杂交分 子,经一定的检测手段将待测核酸在组织、细胞或染色体上的位置显示出来。
原位 PCR 技术首先需要对细胞进行预处理,使细胞具有适当的通透性且保持完整。原位 PCR 的基本原理与普通 PCR 的扩增原理基本相同,通过在处理后的单细胞或组织切片上对特异 的 DNA 或 cDNA 进行原位 PCR 扩增,然后采用 DNA 分子原位杂交技术、免疫组化或荧光 检测技术,通过使用荧光显微镜检测等手段,对细胞内靶片段进行特定核酸序列检出及定位。 原位 PCR 基本原理如图 8.1 所示。
图 8.1 原位 PCR 原理示意图
原位 PCR 可在不破坏细胞的前提下利用原位完整的细胞作为微量反应体系,通过扩增细胞 内的靶片段来进行检测,既有高度的特异性又可精确的定位,综合了 PCR 技术和原位杂交 技术各自的优点,可实现目标物在分子水平上的原位检测和定量研究。但由于取材位置的局 限性,不是所有的靶序列都可以与引物结合而获得扩增,因此为了获得较好的扩增效率,引 物和 DNA 聚合酶的浓度比传统 PCR 要高一些,每一个保温时间都相应地稍长一些。
四、应用及研究进展
自 Hodson 等首次将原位 PCR 应用于原核细胞显示出微生物单细胞内的特定基因和产物在微观尺度上的分布后,原位 PCR 技术由于其精准定位的特性和高度敏感性,在动物学、植物学、病毒学、微生物学、病理学、肿瘤学以及临床医学等研究领域中具有较为广泛的应用。 原位 PCR 被广泛用于鉴定细胞内功能基因的发生及表达,应用于内源基因检测、外源基因 检测、病原体检测、基因突变、基因重排和染色体移位、基因变异的鉴定和基因表达的研究 等方面。
自 Bagasra 成功发展了间接原位 PCR 检测 mRNA 和 DNA 的操作方法,该方法灵敏度和特 异性高,检测时间也大大缩短。实现对目的基因进行组织定位,提高检测诊断的准确性。杨 洪一等利用制备的地高辛标记的特异 cDNA 探针建立了检测辣椒叶组织中辣椒轻斑驳病 毒分布的间接原位 PCR 法。辛丽红等利用原位 PCR 技术检测 56 例肺癌石蜡标本 PTEN 基因 DNA 表达结果表明 PTEN 基因在肺癌组织中的表达较正常肺组织中的表达有所降低, 说明 PTEN 基因的低表达可能在肺癌的发生发展过程中起重要作用,基因的表达与肺癌患 者的性别无显著相关,而与肺癌的病理类型、组织分化程度、临床分期,以及有无淋巴结转 移密切相关。Alzahrani 等将原位 PCR 技术应用于检测肝组织中丙型肝炎病毒以及定位,原 位 PCR 定位结果显示阳性信号主要分布于栅栏组织,其次是海绵组织,表皮组织中也有少 量阳性信号,表明此方法能够对病毒的感染部位进行较为准确的定位。 Nuriya 等建立了一 种高特异性高灵敏度的原位 PCR 方法揭示了乙型肝炎病毒(HBV)和丙型肝炎病毒(HCV) 在慢性肝病患者肝脏中的分布及细胞定位并通过实时荧光 PCR 方法进行结果验证,结果表 明在慢性肝病中,几乎所有的肝细胞都感染了 HBV 或 HCV,这一发现表明,病毒在慢性阶 段在整个肝脏传播。由于原位 PCR 先对待检测基因片断进行扩增,因而具有高度的敏感性, 可检出原位杂交检不出的单拷贝、低拷贝基因。大大提高了这些病原体的检出率。
原位 PCR 技术在动物疾病的检测诊断方面也获得较大的应用。Tereza 等使用标记为生物素 的反向引物对具有超强毒性的传染性法氏囊病病毒巴西毒株(vvIBDV)进行了原位 RT-PCR 检测,与传统 RT-PCR 和病毒分离技术相比具有更高的灵敏度。在寄生虫病原体检测领域, 原位 PCR 技术也有广泛使用。李爽等人利用间接原位 PCR 技术检测鼠感染弓形虫后病原体 在鼠体内各组织器官中的分布。结果显示,弓形虫主要寄生在鼠肝脏、肠道,引起鼠严重的 病理损伤。李爽等用原位 PCR 技术检测动物组织中寄生虫的感染情况。黄广明等应用原位 PCR 对鸡传染性法氏囊早期侵染过程进行了研究。杨素等使用间接原位 RT-PCR 技术对禽流 感病毒进行检测,可检测出约 1μg/L 质粒的 DNA,并能特异性地在细胞涂片和石蜡组织切 片中检测和定位,具有良好的特异性和较高的敏感性。
原位 PCR 在与其他各项分子生物学技术的结合发展中也发挥了较大的优势和作用。Felbel 等将原位 PCR 技术与微流控芯片技术相结合,建立了可诊断细胞群中的癌基因阳性细胞的 技术,对未来医学研究具有重大意义。Raub 等开发了一种结合了微流控区室化、原位 PCR 和荧光显微镜的遗传分析组织镶嵌技术。在结直肠癌组织切片中原位检测 KRAS G12V 突变, 为肿瘤学中的靶向治疗提供了辅助诊断的依据。Gan Wupeng 等将原位 PCR 和微流控装置结 合已成功开发了带有嵌入式壳聚糖改性 Fusion 5 滤纸的塑料微流控装置,用于 DNA 提取和 浓缩,为 DNA 研究提供了新的思路。
为适应定向进化方法改善基因和蛋白质特性的趋势。Shao Weilan 等建立了一种通过降低 DNA 聚合酶的保真度来引入随机突变的原位易出错 PCR 方法(Is epPCR)。此方法通过对 表达质粒中可变 DNA 区域的一步易出错扩增,可以创建和筛选突变体文库;Is epPCR 为通 过定向进化改进基因和蛋白质提供了一种新颖、方便和有效的方法。Hussein 等应用原位 PCR 技术和免疫过氧化物酶染色相结合来检测鸡新城疫病毒的组织趋向性和感染发病机制。原位 PCR 技术与其他技术手段相结合,具有更为广泛的应用。
随着分子生物学及其他学科的发展,各项技术的优势融合将会成为生物技术领域的一个趋势。 原位 PCR 技术与荧光探针定量等多种检测技术相互验证、与微流控装置和生物芯片技术相 结合,原位 PCR 技术作为形态学与分子生物学前沿学科交叉结合的产物对学科前沿的研究 和发展起着重要的作用,随着这一技术的不断改进、完善和发展,原位 PCR 技术将对分子 病理学、分子病毒学、分子肿瘤学以及临床各学科的发展起着很大的推动作用。
第 2 节 原位PCR分类
原位 PCR 技术(In situ PCR,Is PCR )是 Hasse 等人于 1990 年建立的技术,即在组织细 胞里进行 PCR 反应,当时称为“细胞内 PCR”(in cell PCR)。它结合了具有细胞定位能力的 原位杂交技术和高度特异敏感性的 PCR 技术的优点,能检测出细胞内单拷贝及低拷贝的靶 DNA 或 RNA。该方法就是利用完整的细胞作为一个微小的反应体系来扩增细胞内的目的片 段,在不破坏细胞的前提下,利用一些特定的检测手段来检测细胞内的扩增产物。与原位杂 交相比,原位 PCR 技术大大提高了检测的灵敏度。原位技术既可以分辩鉴定带有靶序列的 细胞,又可以标出靶序列在细胞内的位置,对有效检测各种病原菌和从分子及细胞水平上研 究疾病的发病机理和临床转归过程具有重大的实用价值。
原位 PCR 技术作为一种病原体的检测方法,其标记物的类型可分为两种:非放射性标 记法和放射性标记法。非放射性标记法包括如地高辛、生物素、过氧化物酶等,后者放射性 标记法多采用放射性同位素 H3h 和 P32 等。
根据标记物掺入方法的不同,原位 PCR 方法分为两大类:即在反应体系中使用标记的 单核苷酸或引物的直接法原位 PCR(direct in situ PCR);所用引物和单核苷酸都不带任何标记 物,待反应结束后再用原位杂交技术检测特异性扩增片段产物的间接法原位 PCR(indirect in situ PCR)。现在临床上使用最多的是间接法原位 PCR,相对于直接法原位 PCR,它的特异 性更高。另外还有一种叫做原位反转录 PCR 的原位扩增方法是指在进行 PCR 之前首先进行 反转录过程(RT),将反转录的产物作为模板用于扩增,这个过程叫做原位反转录 PCR(In situ reverse transcription PCR,简称原位 RT-PCR)。近年出现的原位环式等温扩增,即在 Is PCR 原理基础上进行了改进,穿透处理条件温和,细胞不易破碎,独特设计的环状引物使扩增产 物分子量大,不易扩散,扩增温度低且恒定,不使用 PCR 仪,细胞损失率大大降低。
一、 直接原位 PCR
直接原位 PCR 法,在进行原位扩增之前,把用同位素、非同位素、生物素或地高辛等 标记的三磷酸核苷酸或引物加入反应体系中,随着核算扩增的进行,标记的核苷酸或引物直 接掺入到产物中,PCR 反应产物用放射性自显影、免疫组织化学反应或荧光检测技术进行 特异核酸序列的检测及定位。直接原位 PCR 具有操作简便、流程短、省时的优点,现已有 不少成功的报道。虽然该法步骤相对较少,可大大缩短时间,但直接原位 PCR 易发生引物
错配或非特异性退火,容易出现假阳性,特异性不如间接法。此外,带有标记的引物还会降 低 PCR 效率。在利用直接法原位 PCR 进行原位扩增时,需注意假阳性的出现,该方法的非 特异性扩增引起的假阳性结果限制了其使用。
直接原位 PCR 技术应用范围广泛,其中之一是能够直接应用光学显微镜观察病原核酸 在组织器官内细胞水平的确切分布和定位,是研究病原致病机理的有效手段。另外,还可以 用于各种动植物流行病毒的检测。2018 年胡敏等提出对甘蔗冰冻切片中甘蔗宿根矮化病 (RSD)致病菌 Lxx(Leisoniaxyli subsp..xyli)进行定位的原位 PCR 方法,利用病原菌 Lxx 16S~23S rDNA 内部转录间隔区(ITS)特异性引物进行原位 PCR 扩增,并对随机参入扩增 片段与地高辛-11-dUTP 进行免疫组化,建立甘蔗样品冰冻切片中直接原位 PCR 方法。结果 表明:直接原位 PCR 对 RSD 蔗株茎样品切片显色为阳性,阳性信号出现于甘蔗茎部的木质 部、韧皮部,而健康蔗株茎样品无信号,方法特异性较好。
二、间接原位 PCR
间接法原位 PCR 实际上是与原位杂交技术的结合,故又称 PCR 原位杂交。它先对细 胞内的目的基因进行原位扩增,再用标记好的探针进行核酸分子原位杂交以定位检测扩增的 目的基因。间接法原位 PCR 可以克服由于 DNA 修复或引物错配引起的非特异性问题,是 目前应用较广泛的靶核苷酸序列原位扩增技术。间接法原位 PCR 体系中的引物和三磷酸核 苷酸都不带标记物。先进行细胞内靶核苷酸序列的原位扩增,然后用原位杂交技术对特异性 扩增的产物进行检测及定位。原位杂交中所用的探针可特异性地与扩增产物的靶序列杂交, 即使扩增产物中有非靶序列成份,它们也不会呈现阳性反应,其特异性比直接法更强。但该 法需制备探针进行原位杂交检测结果,检测时可能会受到探针大小的制约。该方法需要进行 扩增反应后的洗脱和原位杂交过程,因而所需时间相对较长。与直接法原位相比,该方法相 对繁琐,耗时长。
间接原位 PCR 具有灵敏度高、特异性好和实现精确组织定位的众多优点,适用于病 原感染的确诊,病原对不同组织器官的亲嗜性、侵染途径和致病机理等相关研究;同时适用 于动物、植物等内源性基因与外源性基因表达情况的原位检测与分析。2019 年李亚楠等基 于原位 PCR 技术建立了检测牡蛎疱疹病毒(OsHV-1)的间接原位 PCR 方法并进行优化, 通过组织切片上显示的病毒核酸阳性信号,能判别该病毒在不同组织部位的分布特点和受感 染的细胞类型,适用于 OsHV-1 感染的确诊、病毒对不同组织器官的亲嗜性、病毒侵染途径和致病机理等相关研究。
三、原位反转录 PCR
原位反转录 PCR 是指在 PCR 之前首先进行反转录过程(RT),将反转录的产物作为模 板用于扩增,这个过程叫做原位反转录 PCR(in situ reverse transcription PCR,简称原位 RT-PCR)。原位反转录 PCR 原理如图 8.2 所示。
原位反转录 PCR 可用于检测细胞或组织切片中各种病毒 RNA 或 mRNA。李雪梅等根 据猪繁殖与呼吸综合征病毒(PRRSV)的 ORF6 和 ORF7 设计 1 套引物和 37 bp 的寡核苷 酸探针,建立用原位反转录 PCR 检测石蜡组织切片中 PRRSV 核酸的方法,应用原位反转 录 PCR 检测扁桃体、胸腺、脾脏、肺门淋巴结时都出现了阳性信号,阳性对照切片检测到 阳性细胞,阴性对照切片未见蓝紫色颗粒。
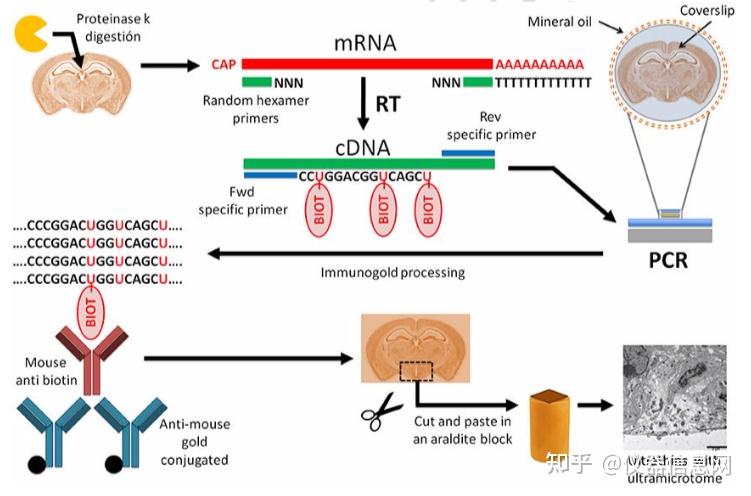
图 8.2 原位反转录 PCR 原理示意图
四、原位环式等温扩增
基于核酸等温扩增 LAMP 技术的性质,原位环式等温扩增 LAMP 技术比传统技术有四 个优点。首先,原位 LAMP 是在一个恒定和相对较低的温度(65°C)下进行的,从而能够产 生可重复的结果,并最大限度地减少对染色体形态的可能损害。引物原位标记技术(cycling primed in situ labeling,C-PRINS)的结果易受到温度和压力变化的影响。在 Meng 等的研究 中,通过 ITS2 的染色体定位来比较 C-PRINS 和原位 LAMP 的性能,由于染色体形态的严 重损伤,C-PRINS 的结果始终不准确。其次,与 Is PCR 不同,原位 LAMP 利用 4 个引物来 扩增,可以实现更高的特异性。原位 LAMP 能够区分高度相似的重复序列,并在不同基因 谱系的染色体定位方面显示出巨大的潜力。第三,原位 LAMP 能产生高分子量扩增产物, 并在相对较低和恒定的反应温度下进行,没有循环变性-退火的步骤,有助于减少扩增产物 扩散的可能。最后,原位 LAMP 不需要制备带有标记的探针,仅需要不超过 1 小时的 2 步 扩增过程,而 Is PCR 不仅需要设计探针,还需要一个过夜(14-22 小时)的扩增过程。原位 环式等温扩增原理如图 8.3 所示。
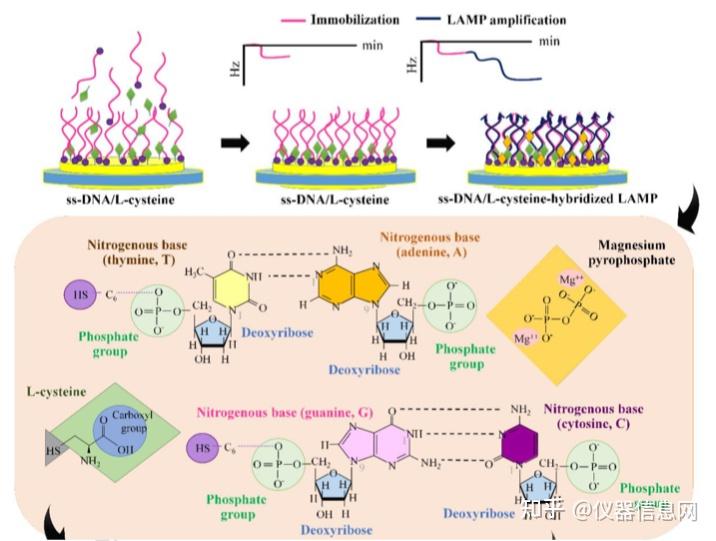
图 8.3 原位环式等温扩增原理示意图
五、原位 PCR 与微流控芯片结合
与传统的热循环仪器相比,低功耗、反应时间快、样品和试剂用量减少是微型 PCR 芯 片的优点。原位 RT-PCR 结合微流控芯片技术在临床诊断上进行了初步应用。Felbel 等将原位 RT-PCR 技术与微流控芯片技术相结合,根据 HPV16 靶癌基因的检测建立了一种应用于 原位 RT-PCR 的肿瘤诊断设备,通过原位 RT-PCR 芯片鉴定细胞群中的癌细胞基因。
第 3 节 原位PCR技术流程
原位 PCR 技术流程可以分为四个关键步骤:细胞固定、蛋白酶消化、原位 PCR 扩增和PCR 扩增产物检测。原位 PCR 技术流程如图 8.4 所示。
一、细胞固定
为了保护细胞的形态结构,需要通过福尔马林、多聚甲醛、丙酮等固定剂来对细胞进行 固定。通过使蛋白质凝集、核酸交联,从而形成有效的限制产物扩散的网络性屏障,以保证 细胞不被破坏。
二、蛋白酶消化
使用醛类固定剂可以通过形成网络性屏障对细胞进行保护,防止扩增产物扩散,但所形 成的形成网络性屏障一定程度上妨碍了 PCR 反应过程中所需试剂进入细胞,难以与靶 DNA 接触。因而需要对固定剂进行消化,能增加细胞的透性有利于使所需试剂与靶 DNA 充分反 应。常用的蛋白酶有胃蛋白酶、蛋白酶 k。消化时间不宜过久,否则容易产生扩增引物扩散 的问题。蛋白消化的强度应该与组织的固定程度相适应。样本消化后,使用的酶要通过加热 去活化或洗脱等方法除去。
三、原位 PCR 扩增
原位 PCR 扩增是在玻片或 Eppendorff 管中进行的。使用引物对细胞内的 DNA 或 RNA 进行选择性扩增。其中对 RNA 进行扩增需要先将其转录成 cDNA 后进行 PCR 循环扩增。 PCR 反应液应含有特异性引物、酶、dNTP 等。反应经过变性、复性和延伸三步。反应体系 中各试剂的浓度和反应时间应根据待扩增片段的不同进行优化。
原位 PCR 扩增的成功应具备以下条件,即足够的扩增效率、避免引物错配、扩增产物 尽可能保留在原位。
四、扩增产物检测
扩增产物可能在细胞间弥散,需要对玻片进行适量振洗,避免背景干扰。洗涤的程度要兼顾减低背景和保留强阳性信号。扩增产物的检测分为直接原位 PCR 和间接原位 PCR。
直接原位 PCR 采用放射自显影、免疫组化、荧光镜检等,即在反应中加入地高辛或生 物素标记的多核苷酸,掺入到扩增产物中,再用地高辛抗体或亲和素进行免疫组化反应来显 示检测结果。使用此方法时应该注意细胞内源性自发荧光的干扰。
间接原位 PCR 不需要标记物直接进行 PCR 反应,反应结束后通过原位杂交技术来检测 扩增信号,此方法可以避免引物错配等引起的非特异性问题。该方法当前使用最为广泛。
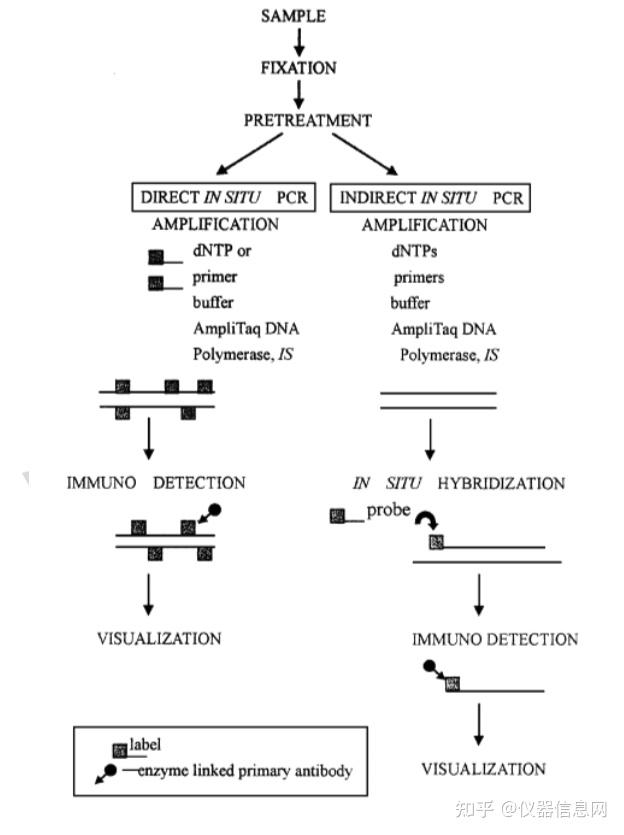
图 8.4 原位 PCR 技术流程图
五、直接原位 PCR 技术流程
(一)试剂耗材与设备
1、主要试剂耗材
固定液:10 %甲醛液
缓冲液 A:0.1mol/L Tris-HCl (pH7.4),0.3% Tween-20,0.5% NP-40
缓冲液 B:0.01mol/L PBS,0.1% Triton-X100
缓冲液 C:0.1mol/L Tris-HCl (pH7.5),0.1mol/LNaCl,2mmol/L MgCl2, 0.5% Triton-X100
PCR 反应缓冲液:两种引物各 100 pmol,dATP、dCTP、dGTP 和 dTTP 各 200μmol,Bio-dUTP 50 mol,1×PCR 缓冲液
蛋白酶 K:20-50 μg/ml,溶于 TE 缓冲液中
Tag 酶:5U/μl
封闭液:3%BSA(溶于缓冲液 C 中)
显色液:0.05 % DAB,溶于 0.05mol/L Tris-HCl (pH7.4)
指甲油(市售)、1%盐酸酒精液、苏木素染色、二甲苯、磷酸缓冲盐溶液(PBS)、湿 盒、
2、主要仪器
显微镜、离心机、原位 PCR 扩增仪。
(二)方法
1、引物设计
通过重组技术与 PCR 合成技术合成所需引物,并使用同位素对引物进行标记。引物长 度一般为 18-24bp 引物间熔融温度的差异不超过 2°C,为增加特异性要求应具有较高的 GC 含量,在试验时根据扩增片段的大小选用一对或多对引物。
2、组织切片和细胞样本的制备。
用 10%的甲醛液固定组织样本 8~48h,使用石蜡包埋后切片(厚度 4μm),将切片置 于新鲜的二甲苯中处理 5min,去除石蜡,放入无水乙醇中浸泡 5min 后取出晾干;对于细胞 样本,使用 PBS 洗涤细胞一次,再加入 10%甲醛固定液静置过夜,用橡胶刷刮下细胞,用 PBS 洗涤细胞两次,吸取 50μl 点在载玻片上。
3、切片预处理(蛋白酶消化)
将干燥后的切片置于 0.1mol/L Tris-HCl 中 5min,浸入缓冲液 A 中,室温孵育 10min; 滴加 20μl 蛋白酶 K 液于标本上,在湿盒中 37°C孵育 20min,常温下,使用 0.1mol/L Tris-HCl 洗涤两次。
4、原位扩增
切片使用 PCR 缓冲液冲洗两次,用滤纸吸去多余缓冲液,置 60°C;每片加入如表 1 的 50μl PCR 反应液,用盖玻片封片,四周封以指甲油。进行原位扩增,原位 PCR 扩增反应 体系见表 8.1,原位 PCR 扩增条件如表 8.2。
原位扩增结束后将玻片浸入无水乙醇中 10min,软化指甲油,除去盖玻片,用缓冲液 B 漂洗两次,用无水乙醇固定 5min,干燥。
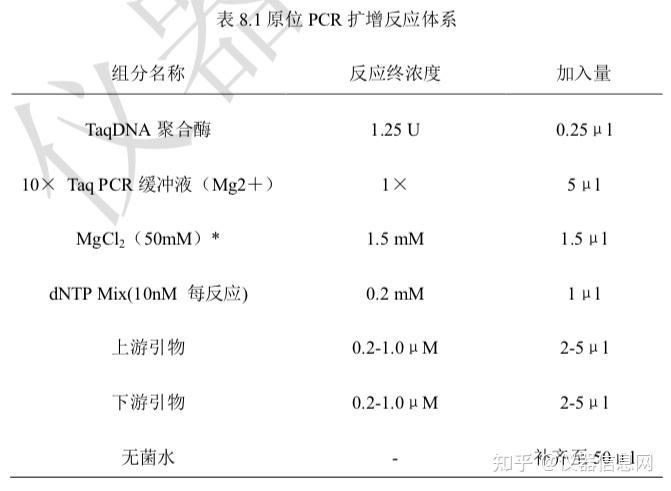
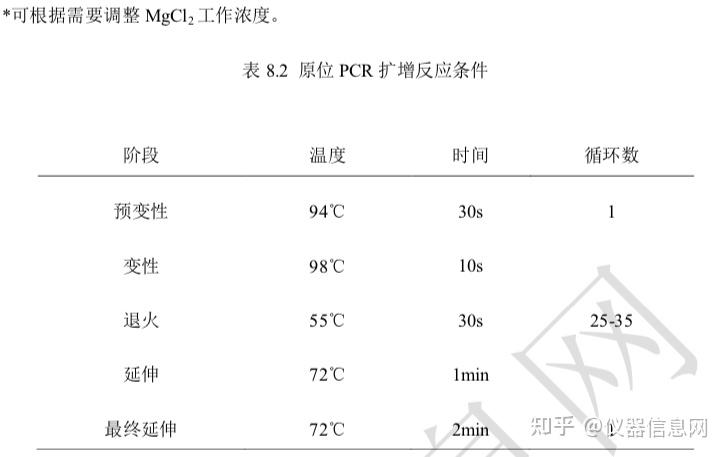
5、检测
在玻片上滴加 30μl 的 3% BSA,封闭静置 1h;用缓冲液 C 冲洗玻片 1min 后,滴加 ABC 缓冲液混合物 20~30μl,室温静置 40min;使用缓冲液对玻片漂洗 15min,重复漂洗 3 次后,滴加显色液于玻片上,镜下观察以控制显色时间(7~14min);流水冲洗玻片后, 置于苏木素液中复染 30s,1%盐酸酒精提抽 2 次;常规脱水、透明、封片,镜下观察结果。
(三)应用
1959 年 Koeh 等,研究了一种引物引导的原位标记法进行 DNA 片段的定位分析。此方法可以看作是直接原位 PCR 的先导。苏俊等研究了一种对石蜡包组织直接原位 PCR 反应 体系和反应条件的优化方法。并对 κ 抗体表达阳性霍奇金淋巴瘤组织中 H/R-S 细胞的 Ig κ 轻链基因重排方式进行了检测。郑宁等采用 5-′非放射性标记引物建立了一种高度特异 的直接原位 PCR 法。对石蜡包埋肝癌( HCC) 及癌周组织中的 HBV DNA 进行了检测。
六、间接原位 PCR 技术流程
(一)试剂耗材与设备
1、主要试剂耗材 2×SSC、50%去离子甲酰胺、Rnase(20μg/ml)、10mmol/L DTT
预杂交液:2×SSC、5%硫酸葡聚糖,50%甲酰胺
杂交液:50%去离子甲酰胺、5×SSC、10%硫酸葡聚糖、5×Denhardt’s 液、2%SDS、 2×SSC、湿盒
2、主要仪器
显微镜、原位 PCR 扩增仪
(二)方法
间接原位 PCR 检测时,靶基因在扩增时,扩增所用的引物和三磷酸核苷酸均不掺入标 记基团,而是使用地高辛标记一段与扩增片段互补的探针。PCR 原位扩增后,再用此探针 进行原位杂交检测扩增产物。本节主要介绍原位杂交,其余方法与直接原位法仅有不在引物 掺入标记这一点不同。
1、预杂交
原位 PCR 扩增后,使用 2×SSC 浸泡后的玻片乙醇脱水;使用 50%去离子甲酰胺 37°C 培养 15 min;每个玻片加入 20μl 预杂交液,培养 30min,2h。
2、杂交
取出培养后的玻片加入 10~20 μl 杂交液,其中包含 0.2~5 μg/ml 标记后的探针,将 盖玻片硅化后覆盖在玻片;使用石蜡膜封片;玻片置于 42°C湿盒中,杂交 12-18 h。
3、杂交后处理
将杂交后的玻片浸入 2×SSC;移去盖玻片,2×SSC 清洗玻片 1~2 min;2×SSC;
使用 2×SSC/50%去离子甲酰胺,42°C清洗 10 min;用 1×SSC/50%去离子甲酰胺,37°C 清洗 30 min,再重复清洗一次;4×SSC/10 mmol/L DTT 清洗 1 h;0.1×SSC 在 37°C下清洗 两次,每次 30 min;在 PBS 中洗 10 min,即可显色;观察结果。
(三)应用
1990 年,Hease 等在组织细胞里进行 PCR 反应,成功检测了真核细胞中 DNA,建立了原位 PCR,使用间接原位 PCR 法成功检测了羊绒毛膜脉络丛细胞中的绵羊脱髓鞘性脑白质 炎病毒,检测灵敏度较直接进行原位杂交高 300 倍。
王剑波等,使用原位间接 PCR 法检测尖锐湿疣组织中 HPV 感染,实验结果表明此方法 具有较高的灵敏度与特异性。李晓等使用免疫组化,原位杂变,PCR 和间接原位 PCR 技术间接 原位 PCR 检测喉鳞癌组织 HPV 感染,并探讨了 HPV 感染与喉鳞癌发生的关系。肖莎等设 计了一对半引物,经过两次 PCR 扩增,建立了半套式间接原位 PCR 技术,成功检测了经 石蜡包埋的非何杰金氏淋巴瘤 bc1-2/JH 融合基因。杨素等通过间接原位 PCR 法成功对细 胞和组织中的禽流感病毒(AIV)进行检测和定位。
七、原位逆转录 PCR 技术流程
(一)试剂耗材与设备
1、主要试剂耗材
DNA 酶、DEPC 水、聚丙烯塑、封条、二甲苯、湿盒 缓冲液:3mol/L NaAc,5μl 1mol/L MgSO4
2、主要设备
原位 PCR 扩增仪
(二)方法
原位逆转录 PCR 主要用于检测细胞内低复制的 mRNA。原位逆转录 PCR 与原位 PCR 的不同有两方面:一是蛋白酶消化后需使用无 RNA 酶的 DNA 酶消化。二是原位逆转录 PCR 胞内的 mRNA 逆转录为 cDNA,再进行 PCR 扩增。此处主要介绍这两个过程,其余方法同原 位 PCR。
1、DNA 酶消化
向预处理后的玻片上加入 1μl 缓冲液、1μlDNA 酶(10U/μl)、8μl DEPC 水;用封 条盖住溶液置于 37°C的湿盒内;消化过夜,去掉封条后用 DEPC 水洗 1min;再用无水乙醇 漂洗;干燥。
2、反转录
在 DNA 酶消化的切片上,加入下列溶液:2μl 25mmol/L MgCl2,1μl RT 缓冲液,1 μl 10mmol/L dNTP,1.5μl DEPC 水,0.5μl 20μmol/L 3’端引物,0.5μlRNA 酶抑制剂,0.5μl 反转录酶;使用指甲油固定封条;在 PCR 仪上 42°C反应 30min;去除封条,使用二 甲苯冲洗 5min 去除指甲油;无水乙醇冲洗 5min,干燥;PCR 扩增,方法同上。
(三)应用
Patel 等利用原位逆转录 PCR 技术检测了人类表皮生长因子受体 mRNA。黄广明等,利 用原位逆转录 PCR 方法研究了鸡传染性法氏囊病病毒 ( IBDV) 的早期侵染过程,且检测 结果较原位杂交灵敏度更高。徐鸣明等,建立了逆转录原位 PCR 检测博尔纳病病毒(BDV) 的方法。 Kher R 等采用限制性酶对样品进行预处理,很好的解决了原位逆转录 PCR 检测 时 DNA 消化不完全的问题。张素娟等利用原位逆转录 PCR 间接法成功检测大肠癌 CD44V6 mRNA 表达。
八、原位 PCR 方法优化
原位 PCR 涉及的技术比较复杂,包括组织学技术、PCR 扩增技术、原位杂交技术、免 疫组织化技术等。原位 PCR 比原位杂交有更高的灵敏度与特异性,HIV 的单拷贝检测可以 有效证明这一点。原位 PCR 还处于相对定量的水平,根据检测出的阳性细胞数进行定量分 析,检出的阳性细胞数与样品中阳性细胞的比例有一定的相关性,实际计数与等量分析仍有 差距。原位 PCR 操作复杂,仍有许多因素影响检测的灵敏度与特异性,需要对反应条件和 反应过程进行优化,提高原位 PCR 方法的灵敏度与特异性。
自 1990 年原位 PCR 被报道以来各个实验室都对此方法进行了研究,使得原位 PCR 技 术日渐成熟,在各个领域应用方面取得了显著的成果。但作为一项发展中的技术仍存在一些 不足,需要进一步优化。
(一)方法灵敏度优化
1、固定液选择:福尔马林长期暴露在空气中会被氧化成甲酸,而 DNA 易被甲酸降解, 在固定细胞时应使用新鲜的福尔马林溶液。利用巢式 RT-PCR 方法分别在新鲜组织、福尔马 林固定、石蜡包埋组织中检测 HCV-RNA,发现石蜡标本的阳性率较新鲜组织低 5 倍,说明 福尔马林固定、石蜡包埋对 RNA 的破坏较大。多聚甲醛或中性甲醛属于交联型固剂,可使 细胞内的大分子凝聚且交联在一起,有利于产物的原位存留。若选用甲醛固定则必须选用蛋 白酶消化,通过乙醇和丙酮固定不宜使用蛋白酶消化,否则会影响灵敏度。
2、固定时间影响:PCR 在扩增石蜡包埋的组织时,组织固定时间越长,DNA 链断裂 基因片段断裂程度越大,扩增后的基因片段越短,灵敏度越高。
3、Taq 酶浓度:玻片会对酶活性产生一定的影响,因此试验使用的酶浓度应稍大一些, 另外小牛血清蛋白可降低玻片对酶的影响,也可使用小牛血清蛋白来减少酶的用量。Mg2 +是维持酶活性的重要物质,浓度为 4.5mol/L 时,阳性信号最强。
4、非放射性标记物浓度: 原位 PCR 中使用 5-溴-脱氧尿嘧啶( Brdu )作为标记物时,显 色信号的强度随着 BdrU 浓度的提高而增强。
5、原位 PCR 反应中,经石蜡包埋的组织切片与新鲜组织相比扩增效率低,易出现假阴 性结果。提高石蜡包埋的组织切片的敏感性,有助于提高方法的灵敏度。
6、PCR 技术具有高灵敏度的特点,相比之下原位 PCR 的灵敏度较低。在原位 PCR 中 对完整的细胞标本上检出单拷贝的 DNA 序列,但在经石蜡等包埋的组织切片上无法检出单 拷贝的 DNA 序列。如何提高石蜡包埋的组织切片灵敏度十分重要。
(二)方法特异性优化
1、RNA 检测:在检测 RNA 前使用 DNA 消化酶消化无关的 DNA。
2、直接原位 PCR 使用经标记的引物进行扩增,PCR 结束后可以直接观察到扩增结果, 操作简单、耗时短,但易发生引物错配或非特异性退火,影响方法的特异性。经标记的引物 会使 PCR 效率降低。间接原位 PCR 是在扩增后使用杂交技术进行检测,很好的解决了引物 错配引起的假阳性问题,具有更高的特异性。
3、消化条件优化:若蛋白酶 K 消化时间过短、酶浓度过低,很难进行有效的扩增, 易出现假阴性。消化时间过长、酶浓度过高,组织细胞的通透性过高,扩增的产物会向细胞 外弥散,导致定位不准确或出现假阴性的情况。在实验前应使用不同浓度蛋白酶 k 和消化时 间进行试验,组织结构模糊的前一个浓度为该组织的最佳消化条件。
4、热启动法提高特异性:Noovo 等创立热启动法,即在 82°C时加入 Taq 酶与引物可以 有效避免引物形成二聚体,提高特异性。或用一种特异性抗 DNA 聚合酶单抗,以阻止引物 非特异性结合。
5、扩增后洗涤:对扩增产物进行彻底洗涤,可降低非特异性背景的影响。
(三)反应条件优化
1、防脱片方法:将盖玻片和载玻片进行硅化防脱片处理放入 2%三乙氧基硅烷(APES) 中浸蘸 1—2s 后干燥。在 4%多聚甲醛对组织切片再固定 1-2h。
2、PCR 反应条件:PCR 扩增中足够的循环数也有利于提高原位 PCR 的灵敏度。但为 防止原位扩增后的产物向细胞外弥散再次扩增导致假阴性结果,循环次数也不宜过多。根据 试验需要,循环次数控制在一个合适的范围。
(四)质量控制
1、设立对照组
为了对原位 PCR 方法进行严格的质量控制,增强结果的可靠性,需要设置多种对照组 保证实验结果的准确性。
阴性对照:1无病毒感染的组织切片,2反应液中无 Taq 酶、引物、或 dNTP。
阳性对照:使用其他检测方法证实的阳性细胞或组织切片。
2、结果判定
根据标记物的性质不同,可分别采用放射自显影、荧光显微镜观察。显色液不同显色结 果也不同。
(1)当加入二氨基联苯胺(DAB),显微镜下观察,受检组织出现棕色沉积物为阳性。
(2)加入四唑硝基蓝(BCIP)氯化硝基四氮唑蓝(NBT),即碱性磷酸酶最佳底物组合之一, 显微镜下观察,受检组织出现蓝色沉淀为阳性。当受检组织不发生显色反应即为阴性。
3、假阴性结果产生质量控制
原位 PCR 中假阴性结果产生可能由固定时间、弥散性伪影、抑制剂、玻片吸收反应物 等产生。
(1)为防止玻片产生的影响,尽量使用与原位 PCR 仪配套的玻片,可以使用多聚赖氨 酸或硅烷处理玻片,防止组织在试验过程中脱落,也可以在反应体系中加入 1%的 BSA 减少 玻片对反应液的吸附。
(2)为了避免产生伪影,可以使用较大的 PCR 产物,并进行彻底的洗涤。
4、假阳性结果产生质量控制
假阳性结果产生可能是由于引物与模板错配、DNA 重组、内源性启动、特异性扩增产 物弥散等。
采用热启动原位 PCR 可以有效减少引物与模板错配。
可以加入一种特异性的抗 DNA 酶单抗,在 70°C以下该抗体与 DNA 酶特异性结合,当温度达到 70°C以上,该抗体被破坏,DNA 酶活性恢复参与 PCR 反应。
内源性 DNA 损伤的 DNA 修复是假阳性结果的常见原因。在组织切片中这种 DNA 损伤后,Taq 聚合酶在修复过程中结合标记核苷酸,可能造成假阳性。 试验中使用不含外核酸酶的 DNA 聚合酶,可以尽量避免 Taq 聚合酶在修复过程产生的假阳性。
使用多对引物扩增,得到长度不同的扩增产物,产物相互交织于原位固定,可有 效防止扩增产物弥散,产生假阳性。
5、其它污染因素控制
原位 PCR 扩增反应都在细胞内完成,反应时生物膜的半透性使引物聚合酶等自由进出 细胞,而扩增产物会被阻隔,这使得污染物难以进入细胞,这些污染物有可能附着于细胞膜外,试验时充分的洗涤可有效去除污染物。
采用专用封闭式试剂盒或使用指甲油在盖玻片四周充分涂匀,防止因盖玻片封闭 不严造成的实验失败。
提高原位杂交的阳性率、降低假阳性结果产生,减少背景因素影响等还是原位 PCR 方 法有待解决的问题。如果能够有效解决原位 PCR 实验中的技术难题,原位 PCR 技术会真 正走出研究实验室,成为诊断实验室的常规手段,在动物学、植物学、病毒学、微生物学、 病理学、肿瘤学以及临床医学等检测和诊断领域中发挥重要作用。
第九章 其他PCR技术进展
第 1 节 反向PCR技术
PCR 技术主要用来快速、大量获得特定已知序列的 DNA 片段。常规的 PCR 技术是用 于扩增两段已知序列之间的 DNA 片段,而对于已知序列侧翼的未知 DNA 序列的扩增,则 毫无办法。但是,在很多时候,人们又非常渴望了解某些特征性遗传标记侧翼的序列,如转 座子或病毒是整合至基因组的什么位点、某一缺失基因丢失了哪一部分、某一 cDNA 的启 动子是什么等。在反向 PCR 技术出现以前,对上述问题的解决办法是:一般首先用 λ 噬菌 体及其衍生物作载体构建相应的基因组文库,接着用杂交的办法获得相应的阳性克隆,然后 用多拷贝质粒再构建次级文库,再进行杂交,最后进行测序。整个过程工程庞大。正是基于 这些原因,TrigliaT 和 OchmanH 等人在常规 PCR 的基础上建立了反向 PCR 技术 (inverse poly-merase chain reaction,IPCR),大大减少了实验的工作量,加快了实验进程。
1、反向 PCR 技术的概念和特点
在已知序列的核心区边侧设计一对反向引物,由于它们的 3’端方向相反,因此不能够 以两引物间的序列为模板形成 PCR 扩增;但当以适当的限制性内切酶裂解含核心区的 DNA, 并用 DNA 连接酶将含有已知序列核心区的 DNA 片段的末端连接形成环状分子,则可以产 生适合于上述两条反向引物 PCR 扩增的模板 DNA,即从两引物分别向未知序列区域延伸到 限制性内切酶切割位点的未知序列。其特点是 PCR 的引物同源于环上核心区的末端序列, 但其方向相反,使链的延伸经过环上的未知区而不是核心区,即扩增的产物含有已知核心区 序列旁边的未知序列。见图 9.1。
图 9.1 IPCR 示意图
2、反向 PCR 的应用进展
(一)用 IPCR 对已知 DNA 区段边侧区域的扩增
反向 PCR 的应用已经证明该方法可以避免不方便的克隆和亚克隆步骤,而获得已知 DNA 区段边侧区域未知序列的 DNA 片段。例如用反向 PCR 扩增 E.coli 天然分离物中转位 插入序列 ISL.的边侧序列;用于编码疟原虫(Plasmodium falciparum)主裂殖子表面抗原前 体的基因;用于获得转录基因的 5'端或 3'端的边侧区域。
(二)用 IPCR 获得启动子序列
在后基因组时代,产生了大量的 EST 及完整的 cDNA 序列,但是由于它们缺乏启动子 等调控序列,因而整个基因序列是不完整的,这对研究基因的表达调控和功能极为不利。通 过 IPCR 来获取启动子可大大降低工作量。但是,用 IPCR 克隆启动子与一般的 IPCR 的差 别在于:它只要克隆已知序列单一方向的外缘片段。因此,对消化基因组 DNA 的限制性酶 的选择就有所不同,该限制性酶的识别位点可位于已知序列上,但不能在引物之间。
(三)用 IPCR 鉴定插入失活基因
在大规模的微生物功能基因组研究中,其中一个很重要的方法是利用转座子随机插入构 建庞大的微生物突变体库,随后对突变体进行功能筛选和表型鉴定,该方法在致病微生物毒 力基因或毒力相关基因研究中是比较常见的,如信号标诱变技术 (signature-tagged mutagenesis,STM)、体内表达技术(in vivo expression technology,IVET) 等。在植物基因组功能研究中,常利用根瘤杆菌的 T-DNA 进行基因的插入失活,然后用 IPC 鉴定相关基因。
第 2 节 巢式PCR
巢式 PCR(nested primers-polymerase chain reaction,NP-PCR)亦 称为嵌合 PCR,通 过设计“外侧”、“内侧”两对引物进行两次 PCR 扩增,外侧引物的互补序列在模板的外侧, 内侧引物的互补序列在同一模板的外侧引物内伽在田一过外伽物扩增含有日的靶序列的较 大 DNA 片段,然后用另一对内侧引物以第一次 PCR 扩增产物(含有内侧引物扩增的靶序 列)为模板扩增,使目的靶序列得到第二次扩增,从而获取目的靶序列。这样两次连续的放 大,明显地提高了 PCR 检测的灵敏度,保证了产物的特异性。对于极其微量的靶序列,应 用巢式 PCR 技术可以获得满意的结果。目前,巢式 PCR 主要可以分为五种:常规巢式 PCR, 半巢式 PCR,反转录巢式 PCR,共有序列巢式 PCR 和特种异巢式 PCR。
1、常规巢式 PCR
巢式 PCR 是一种 PCR 改良模式,它由两轮 PCR 扩增和利用两套引物对所组成,首先 对靶 DNA 进行第一步扩增,然后从第一次反应产物中取出少量作为反应模板进行第二次扩 增,第二次 PCR 引物与第一次反应产物的序列互补。使用常规巢式引物进行连续多轮扩增 可以提高特异性和灵敏度。
巢式 PCR 应用简单,所需条件与常规 PCR 相同,但与常规 PCR 相比,进一步提高了 反应的特异性与灵敏性,在应用中极为广泛。对于用常规 PCR 方法难以扩增出的样品,可 以尝试使用巢式 PCR。
在临床检测中,还会用到单管巢式 PCR 方法,单管巢式 PCR 是在传统巢式 PCR 的基 础上将两对 PCR 引物作特殊的设计,巢式外侧两个引物为 25bp,退火温度比较高(68°C); 巢式内侧两个引物为 17bp,退火温度较低(46°C),PCR 反应液的其它成分与一般 PCR 相 同。这样,通过控制退火温度(68°C)使外侧引物先行扩增,经过 20~30 次循环后(第一次 PCR),再降低退火温度(46°C)使内侧引物以第一次 PCR 产物为模板进行巢式扩增,该 PCR 的灵敏度可达到每毫升样品检出 0.3 个淋球菌,而传统的一步 PCR 法只能检测到 3 个 淋球菌。
2、半巢式 PCR
半巢式 PCR 与巢式 PCR 原埋相同,只是在第 2 轮 PCR 反应中使用的引物有 1 条为第 1 轮 PCR 的引物,这种利用三条引物进行两次 PCR 扩增的方法称为半巢式 PCR (semi-nested PCR),半巢式 PCR 与巢式 PCR 应用条件基本相同。
半巢式 PCR 较巢式 PCR 反应特异性有所降低,易造成非特异产物,可通过提高退火温 度,从常用的 55°C提高为 60°C。从而使模板与引物之间的识别特异性提高,以提高反应的 特异性。半巢式 PCR 多采用第一轮产物直接稀释作为第二轮循环模板,此稀释混合物中的 引物和模板对于第二轮扩增有一定干扰作用,易造成背景高、非特异产物多的缺点,通过对 第一轮产物的初步分离获得纯化的核酸产物,再作为第二轮循环模板可得到更为理想的结果。
当模板 DNA 含量较低时,一次 PCR 难以得到满意的结果,需要进行两轮 PCR 扩增, 与巢式 PCR 相比,在基因的 5'末端或者 3'末端无法设计出两条引物——外引物和内引物, 只能设计出一条引物的情况下,宜采用半巢式 PCR。杜慧等使用半巢式 PCR 成功鉴定出盲 传为阴性的风疹病毒基因型,丰富了我国风疹病毒的基因库;马杰等利用半巢式 PCR 技术 成功鉴定出区域内肾综合征出血热患者的主导基因型。
3、反转录巢式 PCR
反转录巢式 PCR(RT-nested PCR)是在反转录 PCR 的基础上发展起来的,在通过反转 录获得 cDNA 的基础上,对目的基因进行巢式 PCR 扩增。它和简单的反转录 PCR 一样是用 于检测某种 RNA 是否被表达,或者比较其相对表达水平,但是特异性更高、可靠性更强。 通常用于拷贝数较低的 RNA 的扩增,例如扩增丙型肝炎病毒(HCV)感染者体内的 HCV 基因。
反转录巢式 PCR 在分子生物学实验中具有广泛的应用,通常用来获取拷贝数较低的基 因,尤其是在获得 RNA 病毒基因方面。有学者利用反转录巢式 PCR 方法成功获取 HIV-1 主要流行株 Gp120 基因的序列特征,表明该方法是一种检测 HIV 病毒的快速、简便、易行 的方法。黄昆等也利用反转录巢式 PCR 方法扩增了重型血友病患者凝血因子VIII(FVIII)基因 内含子 22 倒位,证明该方法是一种快速准确的内含子 22 倒位检测方法。
4、共有序列巢式 PCR
共有序列巢式 PCR(consensus nested PCR),又称为共有引物巢式 PCR(consensus primer nested PCR),根据同一种属内较为保守的序列,设计简并引物,通常第一轮 PCR 引物的简 并碱基较多,第二轮 PCR 引物的简并碱基较少一些,扩增长度为 200~300bp。引物通常设 计在能够区分微生物的不同亚型的区域内。对于某一种生物,例如病毒,种属内型别很多,但检测样本中的病毒型别又不确定,使用共有序列巢式 PCR 扩增获得目的序列,进而通过 测序获得未知微生物的信息,是一种敏感而又简便易行的检测方法。
共有序列 PCR 常用于检测临床样品中的未知微生物,通过 PCR 扩增获得 DNA 模板, 通过测序以确定未知微生物。Oskar 等利用共有序列巢式 PCR 从白鲷中成功分离出 WBRV 反转录病毒株 A-127/06。Vandevanter 等利用共有序列巢式 PCR 检测了临床样本或细胞培养 中的 21 种疤疹病毒,通过测序发现其中的 14 种是以前没有报道过的。黄欢欢等利用共有序 列巢式 PCR 技术成功证明耐黏菌素菌株及 mcr-1 基因在住院患者及体检人群中均存在一定的流行率,为临床抗感染治疗带来潜在风险。
5、种特异性巢式 PCR
微生物在其分类中有严格的分类方法,属于同一种的微生物在进化中形成了一部分保守 的基因序列,针对种特异的基因设计引物,能够区分同一属内不同种的微生物。在种特异 PCR 中,扩增基因一般选择在较为保守的 16SrRNA 和 23SrRNA 内。近几年来国外学者采 用不同的分子生物学方法对细菌的 16SrRNA 进行研究,得到了广泛的细菌 16SrRNA 的序列 库。现在人们已经认同 16SrRNA/rDNA 基因序列可用于评价生物的遗传多态性和系统发生 关系,在细菌分类学中可作为一个科学可靠的指标。在分类鉴定一些特殊环境下的微生物时, 由于分离培养技术的限制,难以获得它们的纯培养进行生理生化学等指标的分析,这样 16SrRNA/rDNA 序列分析的优越性就体现出来了。
Sturbaum G.D.等利用种特异性巢式 PCR 联合限制性片段长度多态性(RFLP)对一种寄 生虫 Cryptosporidium parum 的卵进行检测。他们设计的引物位于 18S RNA 区域内,通过对 1 个、2 个、3 个、4 个、5 个、7 个和 10 个卵的检测来评价此种 PCR 方法的敏感性,发现 利用 PCR 扩增单个卵基因是可行的,但敏感性需要进一步提高。Matsuki T 等四利用针对于 16S rDNA 的属特异和种特异 PCR 来检测双歧杆菌(bifdobaecteria)。他们提取人排泄物中 的基因组,发现在成年人中链状双歧菌(Bifidobacterium catenulatum)菌群是最常被检测出 来的菌群,在吃奶的孩子中,短双歧菌(Bifidobacterium breve)是最常被检测出来的菌群。 利用针对于 16S rDNA 区的实时定量 PCR 可望检测出排泄物的不同菌群的数量,此种技术 对于检测肠道中的菌群的动态分布提供了很好的办法。Salotra P.等利用针对于莱什曼原虫 (Leishmania donocani)动力体(Kinetoplast)kDNA 的种特异 PCR 检测临床样品中 Leishmania donovani 的感染状况。
第 3 节 多重PCR
多重 PCR (Multiplex polymerase chain reaction,MPCR)是在普通 PCR 的基础上,在同 一个反应体系中加入不同的引物对,针对不同的模板或同一模板的不同区域进行特异性的扩 增,从而得到多个目的片段,结合一定的检测手段进而实现同时对多个靶标进行诊断的技术。 自 Chamberlain 于 1988 年首次提出这个概念以来,MPCR 已经应用于许多领域,包括基因 突变与缺失、基因分型与定量、遗传检测、伴药诊断等。多重 PCR 主要有以下 3 个显著特 点:1高效性,即在同一 PCR 反应管内可同时检出多个基因、多种病原微生物,或对有多 个型别的目的基因进行分型,特别是用非常微量的标本材料就可检测多个基因,大大节省样 本用量。2系统性,多重 PCR 很适宜于成组病原体的检测,如肝炎病毒、肠道致病性细菌、 性病、无芽胞厌氧菌、战伤感染细菌及细菌战剂的同时侦检。3经济简便性,多种基因在同 一反应管内同时检出,大大节省检测时间,节省试剂消耗,降低加样工作强度,节约经费开 支,可更快捷地为临床提供更多诊断信息。
MPCR 所涵盖的范围广,能够同时对多个靶标进行检测,大幅提升检测效率的同时, 还降低了检测成本。随着科学技术的发展,MPCR 技术在扩增和检测方面已经取得了许多新 的突破。在扩增方面,不再局限于在同一反应管中进行扩增,而是将不同的引物对和模板分 散于相对独立的空间中进行扩增;在检测方面,也不断有新的超多重检测技术的出现。目前 多重 PCR 主要可以分为三种:多重荧光定量 PCR,基于微流控的多重 PCR 和基于固相载体 的多重 PCR。
1、多重荧光定量 PCR 技术
(一)基于荧光探针的多重 PCR 技术
多重荧光定量 PCR (Multiple fluorescence quantitative PCR)技术是在荧光定量 PCR 技 术的基础上,利用几种不同荧光基团的组合,结合仪器对不同通道荧光的检测能力实现对多 个靶标的实时定量检测。TaqMan 水解探针 (Hydrolysis probes) 是多重荧光 PCR 体系中常 用的一种探针,探针一端标记荧光基团,另一端标记淬灭基团,通过在不同序列末端标记不 同荧光基团及相应的淬灭基团,即可形成不同的 TaqMan 水解探针,将上述探针及相应的扩 增引物加至同一反应体系,即可实现对多个靶标的共同检测。目前已成功在单管中实现两重 、 三重 PCR 扩增和检测体系,并实现对部分病毒样本的扩增。分子信标(Molecular beacon) 是多重 PCR 体系中另一种常用的探针,当体系中不存在特异性的靶标时,分子信标会自发 形成茎环结构,淬灭基团与荧光基团相互接近从而发生 FRET,不会产生荧光。如果体系中 存在特异性的靶标,在一定的条件下,茎环结构会打开并且与靶标进行复性,从而产生荧光 信号。那么在同一个反应体系中加入几种靶标特异性的分子信标就能够实现对多个靶标的同 时检测。目前基于分子信标已实现了针对不同的靶标的四重、五重 PCR 的检测。
(二)基于探针编码的多重 PCR技术
基于水解探针和分子信标探针的多重荧光定量 PCR 技术的优点是简便快捷,然而受仪 器荧光检测通道的限制,往往最多只能达到四重或五重检测。组合探针编码(Combination probe coding)技术的开发,使得目前的多重实时核酸扩增可检测的靶标数目多于仪器可检 测的数目。通过对 4 种不同的荧光基团进行相互组合可以形成 15 种指示探针,在多重反应 体系中加入多种靶序列特异的置换探针(Displacing probes),那么在检测通道有限的情况 下仍能够实现对多种不同的靶标同时进行扩增和检测。Huang 等利用多色组合探针编码技术 (MCPC)实现了对 8 种食源性病原菌的鉴定以及通过 4 色组合探针编码技术实现了对 15 种 不同型别的 HPV 病毒进行分型。
图 9.2 荧光元素之间的组合
(三)基于荧光染料的多重 PCR技术
非特异性的荧光染料嵌入到 DNA双链小沟中后,在特定的激发光激发后将会产生一定 波长的荧光。以此为基础发展了熔解曲线(Melting curve)分析法(图9.3)。Singh等利用 多重实时 PCR熔解曲线分析的方法,成功地实现了同时对氨苄西林、链霉素、磺胺、四环 素、氯霉素耐药的沙门氏菌的检测与辨别。针对不同的型别设计了特异性的引物对,从而对 模板进行扩增生成不同的扩增子,扩增子之间Tm值各有差异,最后通过熔解曲线峰的位置 进行鉴别。在熔解曲线分析技术的基础上发展而来的高分辨率熔解曲线技术是一种利用饱和 染料,借助高分辨率的仪器,实现对单个核苷酸熔解温度不同从而形成不同形态熔解曲线,进而对基因进行分析的新技术,它具有极高的灵敏度,能够检测出单个碱基之间的差别,目 前主要应用于单核苷酸多态性分析、甲基化分析、基因突变、基因分型等领域。
图 9.3多重熔解曲线
(四)基于荧光探针熔解曲线的多重 PCR技术
由于荧光基团本身发射的不是单一波长的荧光,而且PCR仪对不同荧光基团发射的荧光 信号的区分度有限,所以现有的 PCR仪只能检测4–5种不同的荧光信号。而基于荧光染料的 多重PCR技术由于染料不具备特异性的识别能力,所以同一反应体系中通常只能使用一种荧 光染料。为了弥补这两种方法的不足而开发出来的荧光探针熔解曲线技术(Fluorescence probe melting curve technique)具有成本低、多重检测能力更强的优点。通过在靶序列中选 取相对保守的区域设计通用扩增引物,然后在引物对之间选取差异度较大的区域设计能够识 别各种目标核酸序列的特异性探针,探针在扩增的过程中不会被完全水解,并且根据检测通 道的不同将探针进行分组,每个通道中包含多个特异性探针,通过调整探针的长度或GC含 量对每条探针的Tm值进行人为的调控,使得同一检测通道内针对不同靶标的检测探针Tm值 都间隔合适的差值,每个荧光通道均可定性检测多个靶标,最后通过熔解曲线进行分析。 Liao等利用荧光探针熔解曲线技术成功地实现了对15种高危型HPV的分型以及实现了对48 种人类单核苷酸多态性的分型和分析。
2、基于微流控的多重 PCR技术
基于微流控芯片 (Microfluidic chip)的多重 PCR 技术通过在芯片微孔中预装填不同的引 物对的方法将各重扩增限制在各自的独立空间内,再通过液流控制系统将模板引流到不同的 微孔中,进而在微孔中进行特异的 PCR 扩增反应,最后再进行终点定性检测(图 9.4),从 而实现对多个靶标的区分和鉴别,该方法可有效的避免各核苷酸链之间形成二聚体甚至多聚体所导致非特异性扩增的出现,扩增效率下降,检测灵敏度和特异性降低,最终造成检测结 果假阳性或假阴性。部分研究者结合微流控技术研发了一种可以同时检测多种病原体的诊断 平台,称为“FilmArray”,该平台成功地实现了在 1h 内对 21 种常见的病毒或细菌的检测和 鉴别。有学者设计的一种微流控芯片利用双向电泳技术和多重阵列 PCR 终点荧光检测技术 实现了在全血中对不同病原菌的即时检测。多重液滴式数字 PCR 的作用原理与基于微流控 芯片的多重 PCR 的原理相似,通过芯片装置和液流控制系统产生成千上万个液滴,每个液 滴中只包含单个目的 DNA 分子,这样就可以使不同的靶标处于相对独立的空间进行各自特 异性的扩增反应,最后通过计算各靶标发生有效扩增的液滴数,从而实现绝对定量。 Jackson 等利用液滴式数字 PCR 成功地实现了对血液游离 DNA (Free DNA)中罕见癌症突变的多 重检测,利用液滴式数字 PCR 对珍贵的临床标本进行多靶标的检测,极大地提高了标本的 利用率并降低了成本。基于微流控芯片的多重 PCR 技术和多重液滴式数字 PCR 的优点是在 一定程度上避免了非特异性扩增产物的生成以及在荧光定量 PCR 的基础上增加了检测重数。 但是当模板浓度较低时,基于微流控的方法将模板分流至含特异性引物微孔中的概率降低, 易产生假阴性结果;而多重液滴式数字 PCR 的方法使每个液滴中仅包含单重引物和模板的 概率相对较低,往往一个液滴中包含多对引物或者液滴中的引物与模板为非特异性配对,另 外它对模板的要求较高,浓度过高的模板不能保证生成的每个液滴中仅包含单个目的 DNA 分子。此外在检测方面,上述两种技术都需要配合扩增的特殊性而使用到高精密度的检测仪 器,其技术的复杂性使其实现较为困难。
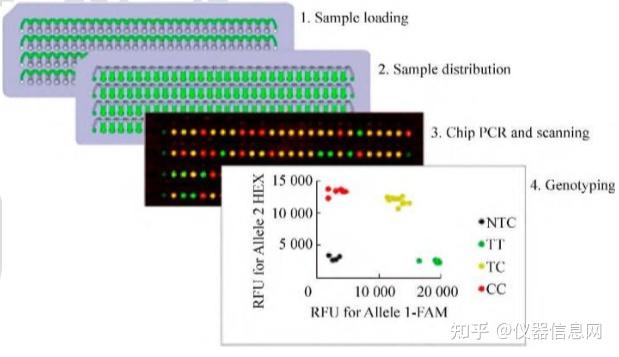
图 9.4 基于微流控的多重 PCR
3、 基于固相载体的多重 PCR技术
(一)基于膜层析的多重检测技术
膜层析技术 (Membrane chromatography) 是在柱层析技术上发展而来的一项技术,以膜介质为基质,偶联相应的离子交换、亲和、疏水等化学基团,制作而成的一种新型的层析介 质。膜层析技术具有简单快速并且能够自动分离液体混合物的优点,而多重PCR技术具有有 效富集目的片段的优点,将这两种技术有机地结合在一起能够形成一种灵敏度高、特异性好、 成本低、简便快速的多重核酸检测方法(图9.5)。基于胶体金标记的核酸探针和侧流试纸条 的基因方法,能够在15 min内检测出核酸样本。侧流微阵列方法能够快速、灵敏地对多种核 酸进行检测,并且不需要专门的仪器设备,这种方法以微型侧流酸检测试纸条在现场实现了 对108例献血者血型相关基因型的准确鉴定。基于膜层析的多重检测技术以其操作简单、携 带便捷等优点拓宽了可检测的范围,使检测不再局限于实验室,对于现场检测也能够实现, 但是膜层析技术中样品的阻滞、非特异性杂交等问题仍然是今后需要解决的问题。
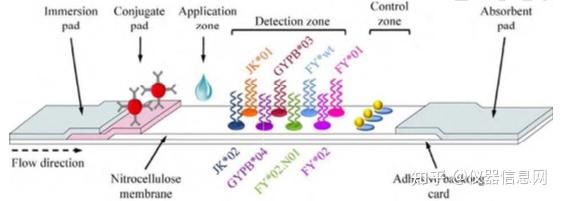
图9.5 基于膜层析的多重检测
(二) 基于Luminex液相芯片的多重检测技术
液相芯片技术 (Liquid chip technology)被称为后基因组时代的芯片技术,也被称为 xMAP技术。它可以同时对1份标本中的100种不同目的分子进行检测,并能在30 min内检测 96个不同的样本。其液相微球编码原理为,将两种荧光染料分别用10种不同的浓度两两混合 形成10×10的荧光配比阵列,并对微球进行染色,得到100种不同荧光编码的微球。将荧光 编码且共价结合了不同捕获探针的微球悬浮于一个液相体系中,先加入待检测分子与其反应, 再加入带有荧光标记的报告分子,从而形成“微球-捕获探针-靶标-报告分子”复合物(图9.6)。 检测时,该复合物在鞘液的作用下将逐个通过检测通道,接受两束不同波长的激发光照射并 对相应的发射光进行检测,其中一束激光用于识别微球种类,即判定该种微球是对哪种特定 的靶标进行检测,另一束激光用于检测微球上报告分子的荧光强度,从而实现对待测物质的 定性和定量分析。目前,Luminex液相芯片技术已广泛的应用于各类病原微生物的检测和分 型,包括对沙门氏菌的检测和分型,禽流感病毒、传染性猴气管炎病毒和空肠弯曲菌的检测和鉴别等。虽然基于 Luminex液相芯片技术能够实现同时对多种不同病原体的检测和鉴别, 但它仅在检测方面发挥了其多通量、高效率的特性,其实质上并没有解决多重扩增中众多引 物、探针、模板间相互错配的问题。因此,未来开发出微球表面多重扩增以及 Luminex液 相芯片检测一体化的技术是核酸多重检测的重要研究方向。
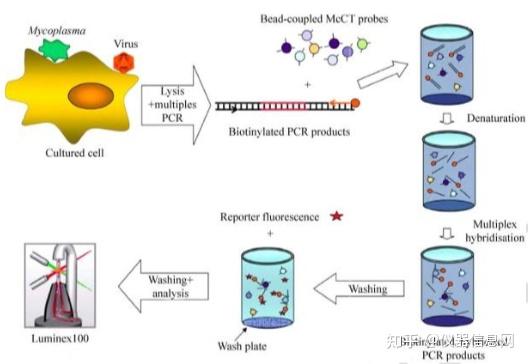
图 9.6 基于 Luminex液相芯片的多重检测技术
第十章 PCR仪校准溯源

第 1 节 PCR仪计量特性


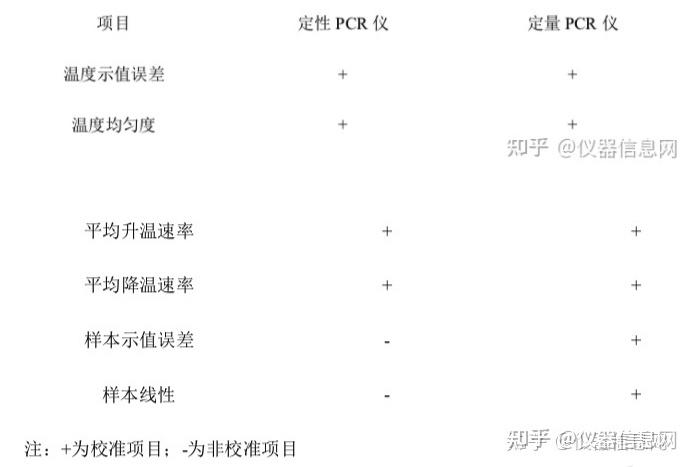
表 10.1 参照 JJF1527-2015《聚合酶链反应分析仪校准规范》的 PCR 仪计量特性

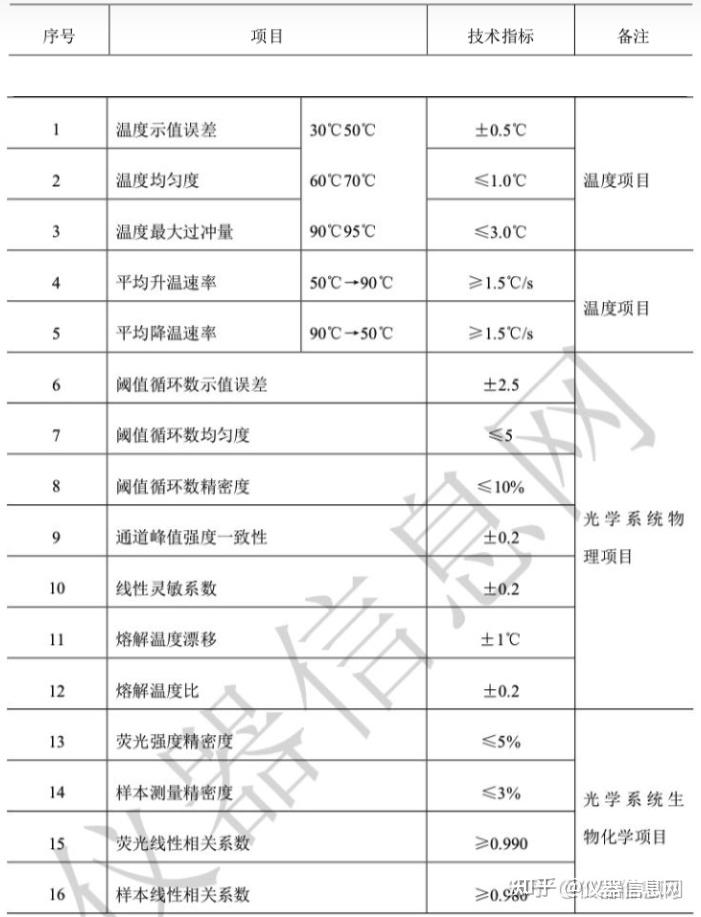
表 10.2 参照 JJF(津)04-2020《实时荧光定量 PCR 仪校准规范》的 PCR 仪计量特性

第 2 节 PCR 仪的期间核查




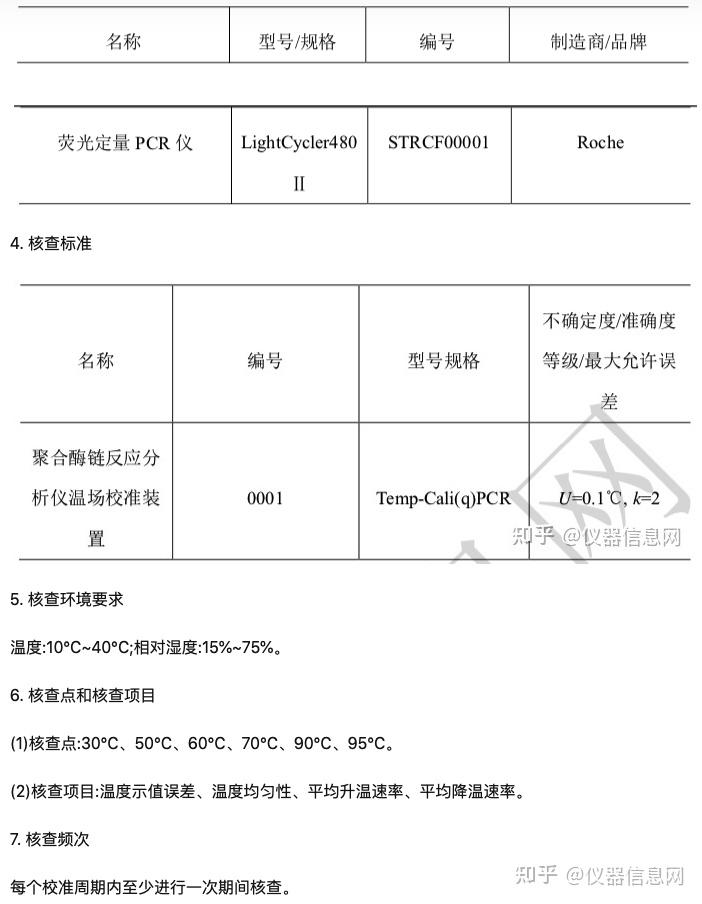

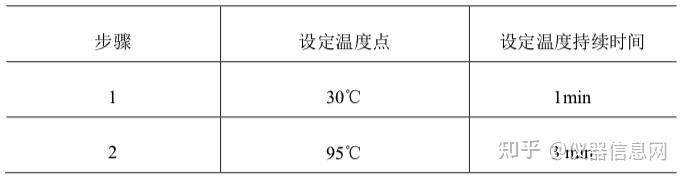
(1)温度示值误差按公式(1)计算:



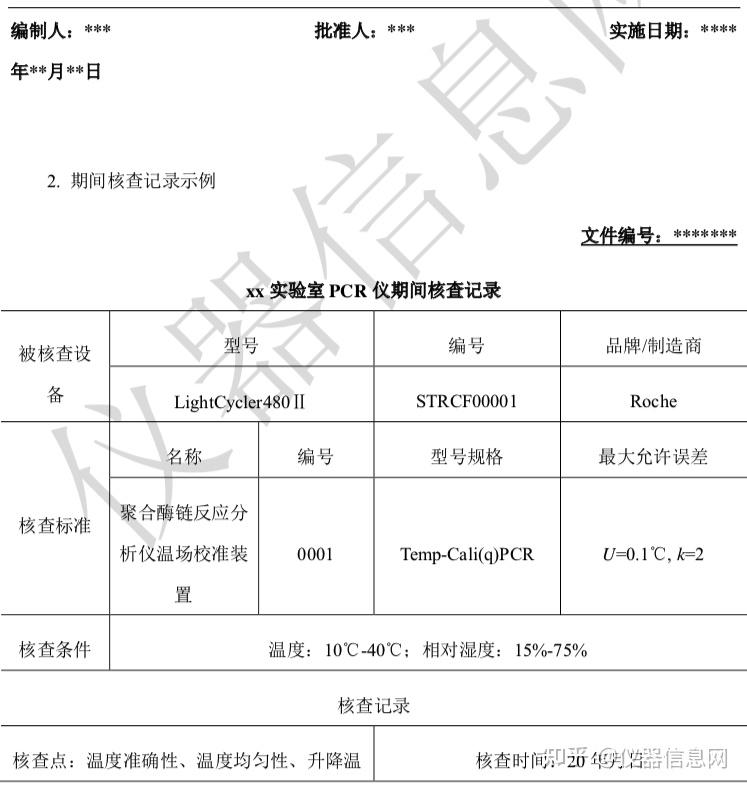
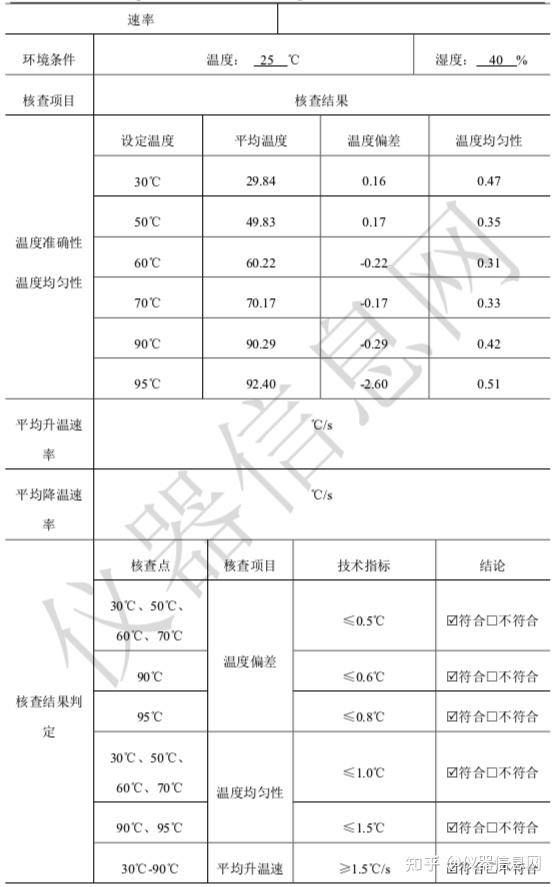
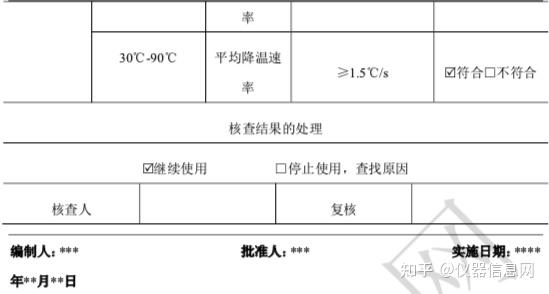
(2)不具备温场检测装置的 PCR 仪期间核查规程示例
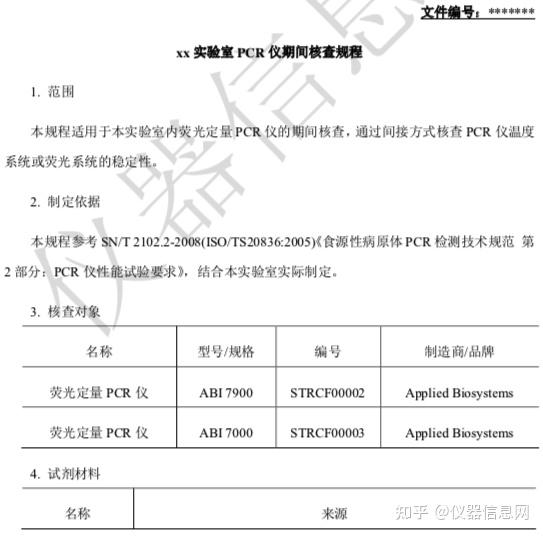
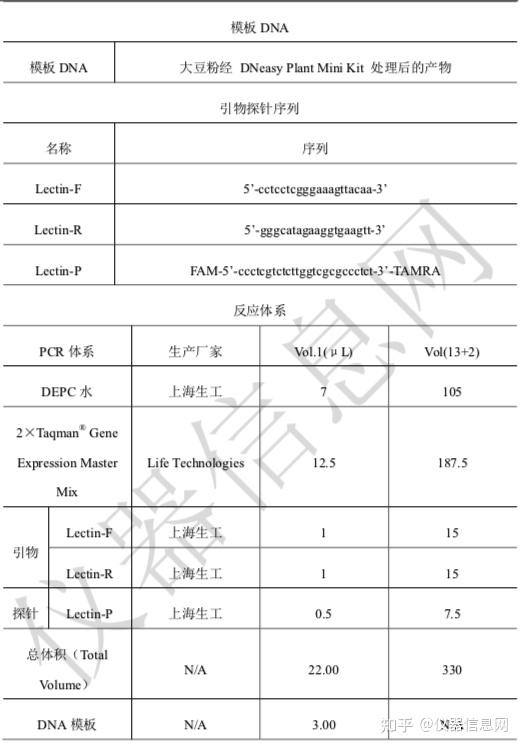


(2)核查结果的处理
若核查结果符合要求,可继续使用;
若核查的检测结果接近临界要求值时,应加大核查 频次或采取其他有效措施(如校准)做进一步验证以规避风险。

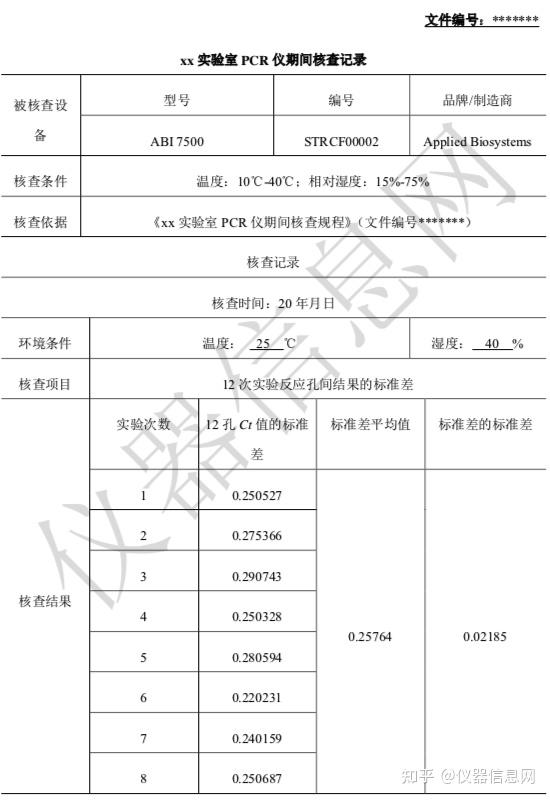
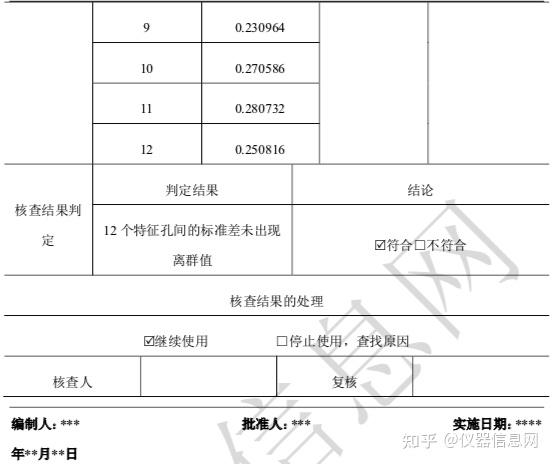



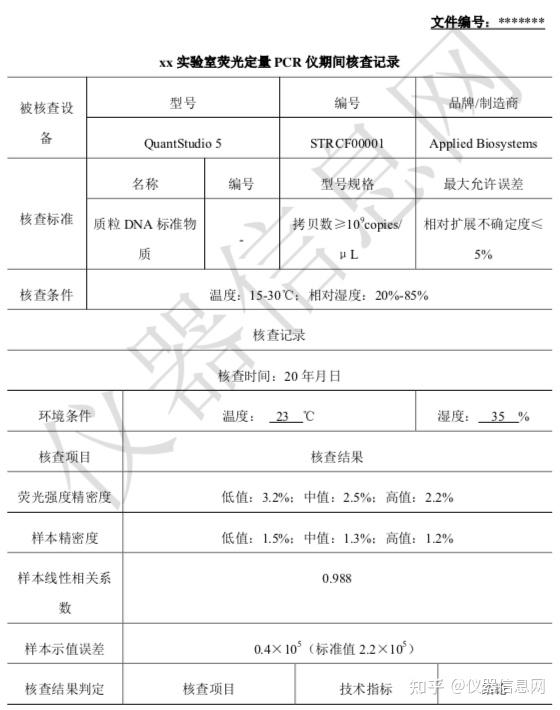
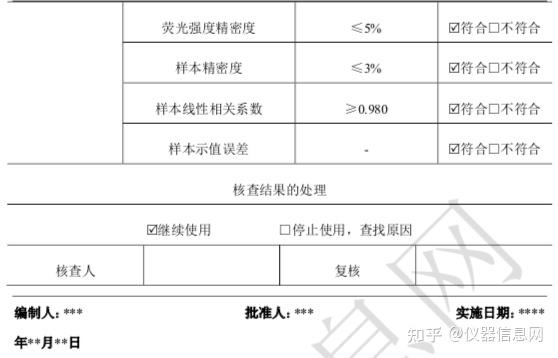
第 3 节 PCR仪的校准



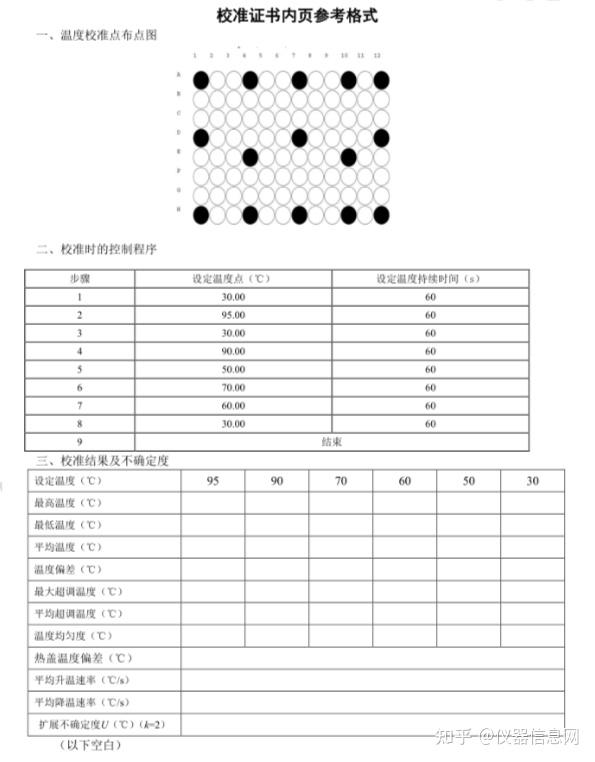
图 10.1. PCR 校准证书内页示例


第十一章 常见问题及解答
第 1 节 PCR 技术及应用

第 2 节 PCR 实验室要求及防污染措施

第 3 节 PCR 操作及判读




进阶版实战宝典(内容更丰富)内容下载:
相关阅读
<hr/>致歉,由于知乎对文章的字数要求不能超过10万字,所以最后两章只能用图片的方式来呈现。
码字不易,感谢您的点赞和收藏~
原文地址:https://zhuanlan.zhihu.com/p/494966197 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-19 08:10
发表于 2024-9-19 08:10



