Part.1 一、Western blot 技术起源
1975年,Edwin Southern发明了DNA杂交检测技术,命名为Southern blot。1977年,James Alwine、David Kemp和George Stark发明了RNA杂交检测技术,命名为Northern blot。两年后位于美国斯坦福大学的George Stark发明了将蛋白从电泳凝胶上转移到活化纤维素上的技术,随后Harry Towbin发展出更快速、简单的电转技术。1981年Neal Burnette正式将这一技术命名为“Western blot”。 Part.2 Western blot 用来干什么? (一)确定目的蛋白的表达情况
这是Western blot最常规的应用,如检测患者的标本与正常人的标本、实验条件处理后细胞与未处理的对照细胞之间目的蛋白的表达变化情况,通过灰度分析可以确定目的蛋白表达水平是升高还是降低。 (二)研究蛋白间的相互作用
这是与免疫共沉淀(co-immunoprecipitation,CO-IP)技术相结合的一种检测蛋白质之间相互作用的常用方法。通过CO-IP技术将相互作用的蛋白质复合体分离出来后进行凝胶电泳,然后利用相互作用的蛋白质的特异性抗体进行检测。适用于对已知的蛋白质的相互作用进行检测,不适用于检测一个蛋白质未知的相互作用蛋白;也可以用于蛋白质-DNA、蛋白质-RNA相互作用的后续分析。 (三)确定目的蛋白的细胞定位
通过分别提取细胞不同部位的蛋白质,如膜蛋白、胞浆蛋白核蛋白或线粒体蛋白等,将分离出来的不同部位的蛋白质进行Western blot检测可以分别检测目的蛋白在不同部位的表达情况。 Part.3 Western blot实验原理
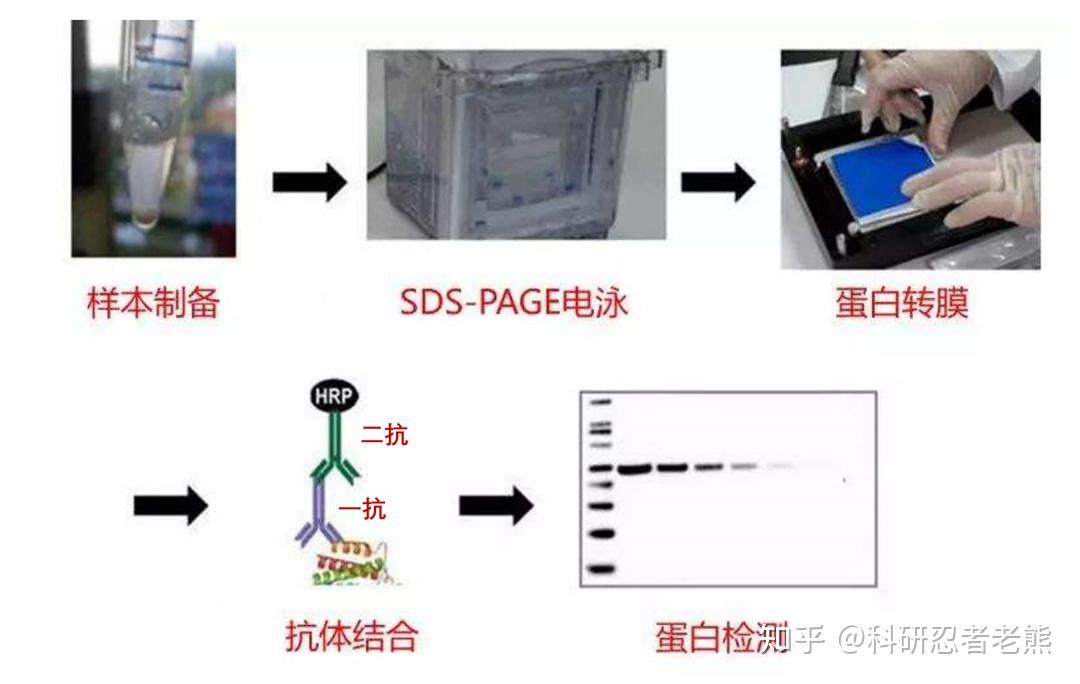
Western blot采用的是聚丙烯酰胺凝胶电泳,通过跑蛋白凝胶电泳,分离蛋白质样品,然后将凝胶转移到某种固相载体(如PVDF膜)。这个固相载体能以非共价键形式吸附蛋白质,同时能保持电泳分离的多肽类型及其生物学活性不变。以载体上的蛋白质或多肽作为抗原,在载体上孵育上对应的抗体进行免疫反应,然后再孵育已经标记好酶的二抗,经过底物显色来检测电泳分离的目的基因表达的特异蛋白成分。
https://www.yeasen.com/news/detail/73
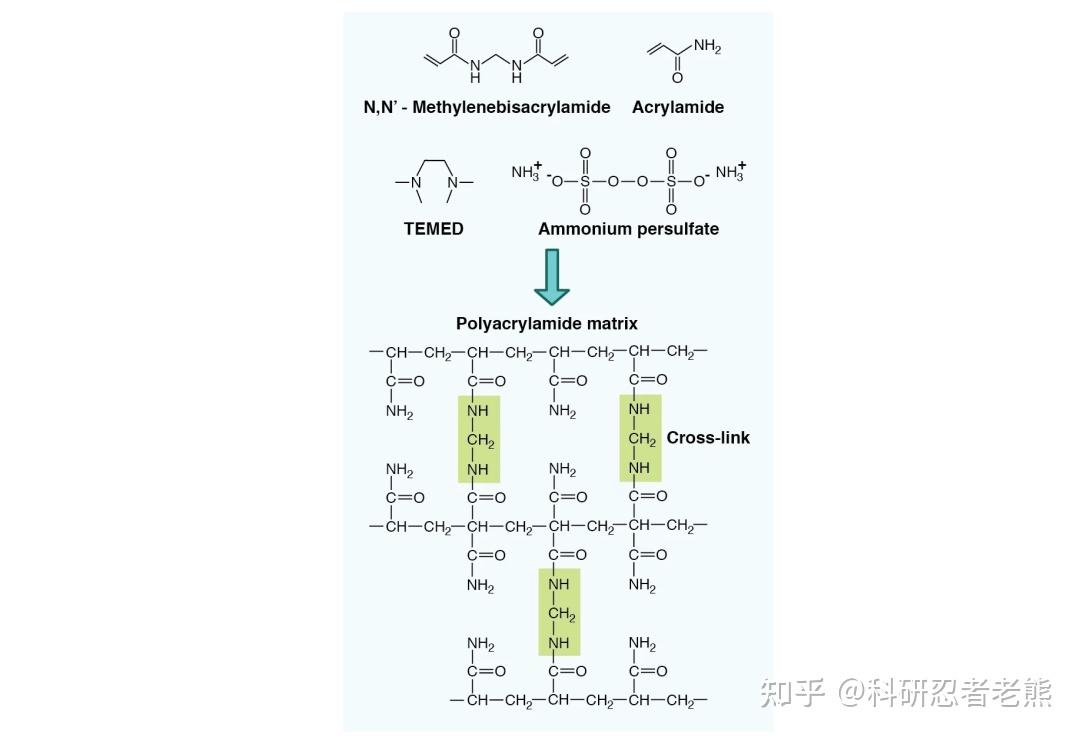
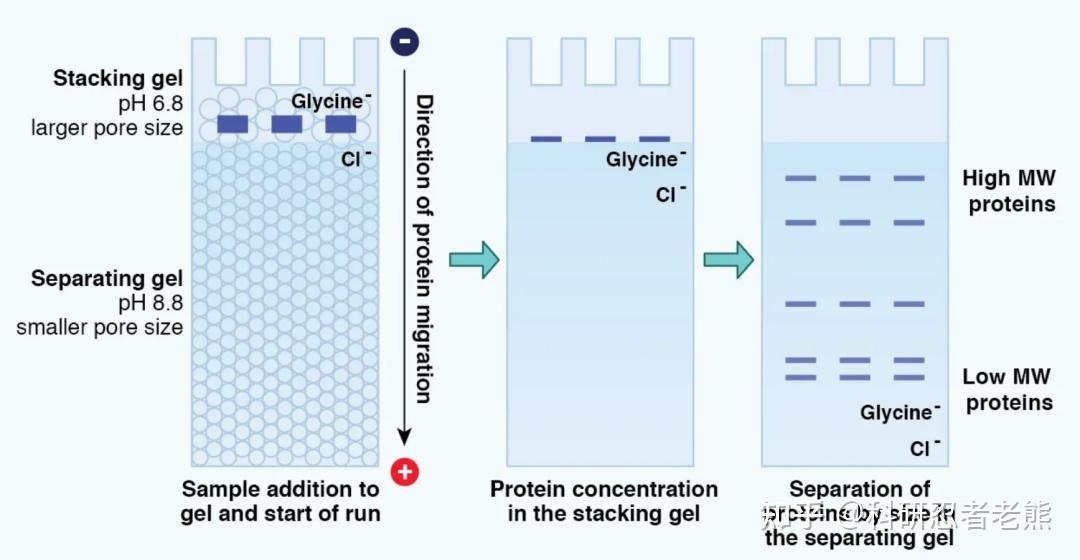
我们平时说的跑蛋白胶一般就是指聚丙烯酰胺凝胶电泳,简称为PAGE(polyacrylamide gel electrophoresis),聚丙烯酰胺凝胶为网状结构,具有分子筛效应,有两种形式:非变性电泳(Native-PAGE)和变性电泳(SDS-PAGE)。非变性电泳(Native-PAGE),是在不加入SDS和巯基乙醇等变性剂的条件下,对保持活性的蛋白质进行聚丙烯酰胺凝胶电泳,常用于同工酶的鉴定和提纯。未加SDS的天然聚丙烯酰胺凝胶电泳可以使生物大分子在电泳过程中保持其天然的形状和电荷,它们的分离是依据其电泳迁移率的不同和凝胶的分子筛作用。非变性电泳分离后仍能保持蛋白质和酶等生物大分子的生物活性,对生物大分子的鉴定有重要意义。在电泳的过程中,蛋白质能够保持完整状态,并依据蛋白质的分子量大小、形状及其所附带的电荷量而逐渐呈梯度分开。
Polyacrylamide Gel Electrophoresis PowerPoint Presentation, free download - ID:345406 (slideserve.com)

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-9-18 22:07
发表于 2024-9-18 22:07












 提升卡
提升卡


