| 通用类别 | 初始事件和环境示例 |
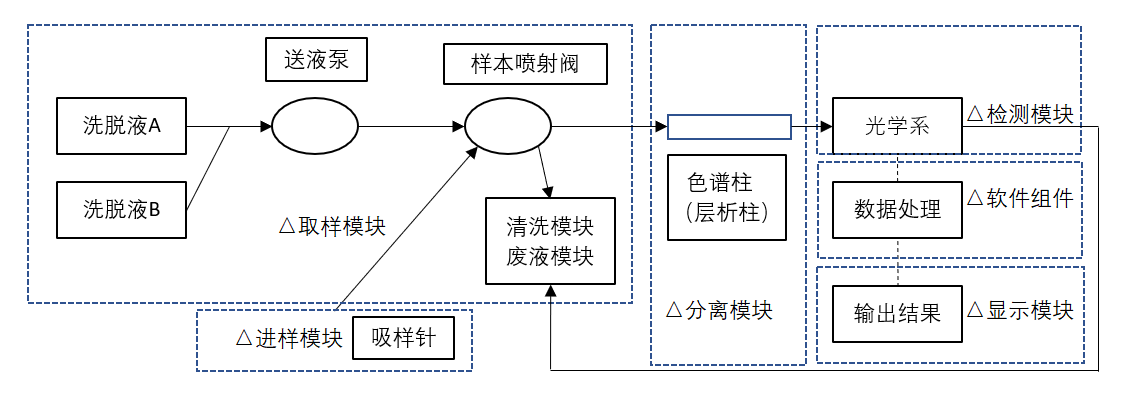
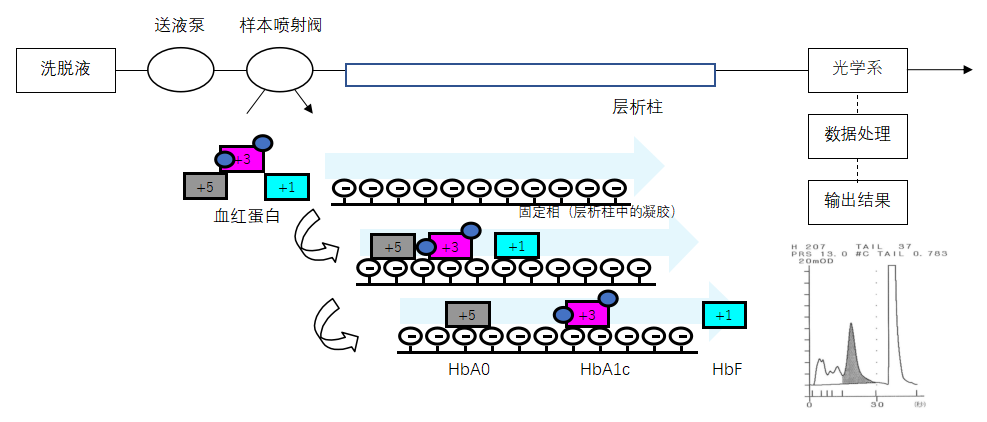
| 不完整的要求 | 设计参数的不恰当规范:可触及金属部分、外壳等与带电部分隔离/保护设计缺陷,导致电击危险防护较低,可能对使用者造成电击危害;设备插头的插销从内部电容器接收电荷过高;工作台支撑件强度不足,设备面、角、边粗糙,可能对使用者造成机械损伤;受潮防护能力不足,造成电击危害;运动部件功能失效,造成机械危害;电磁兼容性不符合要求,导致设备自身不能正常工作或干扰其他设备的正常工作;管道漏液、堵塞和设备浸水,导致电击危害。性能参数不恰当规范:准确性、重复性等不符合要求。机械模块失效:无法进样、吸样量不准确、传送错误、定位不准确,导致检测结果异常。与废液处理相关的生物安全性问题。服务中的要求不恰当规范:说明书未对糖化血红蛋白分析仪的使用方法、设备的维护、保养进行说明。元器件、附件或组件功能失效:显示故障、打印故障和配件错误,导致设备无法正常工作,安全性能出现隐患。 |
| 制造过程 | 控制程序(包括软件)修改未经验证,导致产品的性能参数不符合要求。生产过程中关键工序控制点未进行检测,导致部件、整机不合格。供方的控制不充分:外购件、外协件供方选择不当,外购件、外协件未进行有效进货检验等。 |
| 运输和贮存 | 不适当的包装:产品防护不当导致设备运输过程中损坏。不恰当的环境条件等:在超出设备规定的贮存环境(温度、湿度、大气压力)要求下,导致设备不能正常工作。 |
| 环境因素 | 物理学的(如热、压力、时间):过冷、过热的环境可能导致设备不能正常工作;未对使用环境的条件进行严格验证,以致使用条件的不适应而导致检查结果不准确。电磁场(如对电磁干扰的敏感度):抗电磁干扰能力差,特定环境中设备工作不正常;A类设备在B类设备的环境中使用会对公共电网产生影响,干扰公共电网中其他用电设备的正常运行。不适当的能量供应:设备的供电电压不稳定,导致设备不能正常工作或损坏、检查结果不准确。 |
| 清洁、消毒和灭菌 | 未对冲洗、清洁过程进行确认或确认程序不规范。使用者未按要求进行冲洗、清洁等。 |
| 处置和废弃 | 没提供信息或提供信息不充分:未提供产品使用后处置或提示不充分等。 |
| 人为因素 | 设计缺陷引发的使用错误等。——易混淆的使用说明书如缺少详细的使用方法、缺少必要的技术参数、缺少必要的警告说明、缺少运输和贮存环境条件的限制;设备在故障状态(如变压器过载、断开保护接地线、设备的元器件出现故障)下运行可产生危险警示不足;使用前未检查设备工作状态;操作说明过于复杂,不易懂;未说明如何正确维护、保养设备/附件。冲洗、清洁程序不明确或不清晰。设置或测量参数未标示单位。样本的生化危害性,人员防护问题。控制与操作不对应,显示信息与实际状态不对应。由缺乏技术的/未经培训的人员使用:使用者/操作者未经培训或培训不足,不能正确使用和维护、保养设备。 |
| 失效模式 | 由于老化、磨损和重复使用而导致功能退化/疲劳失效等。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-18 15:49
发表于 2024-9-18 15:49