金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
为了更好的理解基因编辑,我们先补充一些最基础的遗传学知识和背景。
19世纪末,孟德尔对豌豆杂交试验的后代进行观察和统计分析,发现了基因的分离定律和自由组合定律,开创了遗传学。
1910年以后,摩尔根又以果蝇为实验材料,分析遗传连锁现象,证明基因在染色体上呈线性排列,提出了遗传连锁定律,进一步发展为细胞遗传学。
1941年,比德尔以红色面包霉为材料,进行生理生化功能研究,发现基因通过酶起作用,提出“一个基因一个酶”的理论。
20世纪50年代前后,格里菲斯通过肺炎双球菌转化实验,赫尔希和蔡斯通过噬菌体侵染细菌实验,证明了遗传物质为DNA而非蛋白质。
1953年,沃森和克里克通过分析DNA的X射线衍射图谱,提出DNA分子双螺旋结构模型,遗传学进入分子遗传学时代。
20世纪70年代末就有研究组发现,外源DNA可以被酵母或细菌吸收并随机整合到基因组中。随后的工作表明,这个过程也可以以一种有针对性的方式发生,将DNA微注射到细胞核中可以刺激细胞同源重组,从而实现有针对性的基因组修饰。
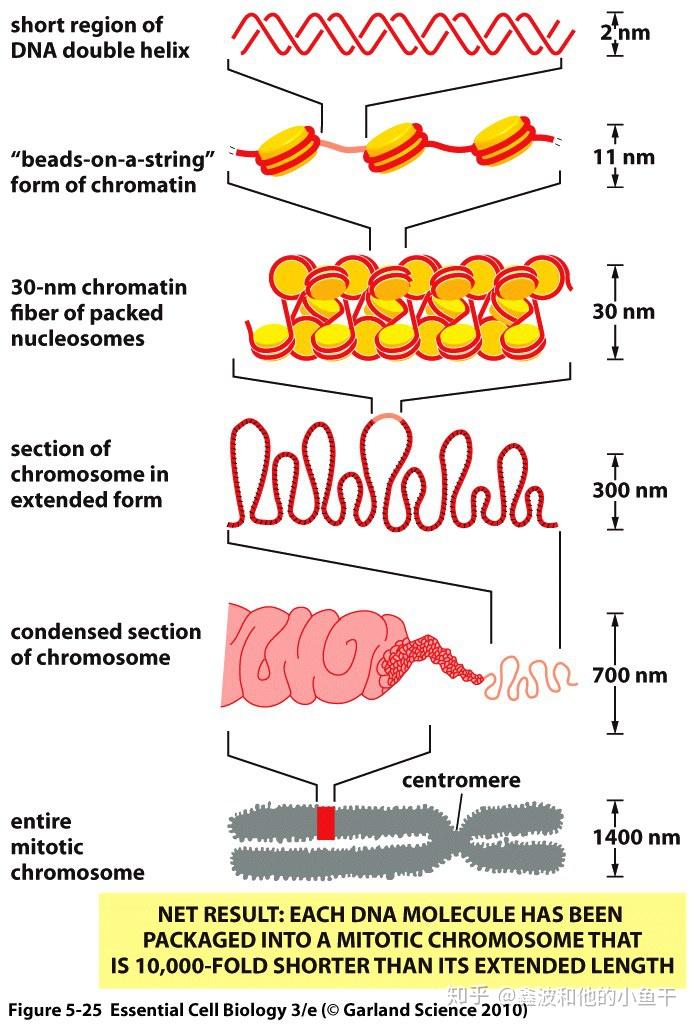
DNA缠绕在组蛋白上形成核小体,经过缠绕折叠变得密集,形成染色体。染色质和染色体是同种物质在不同细胞时期下的不同状态,分裂期染色质浓缩成棒状染色体以便于分派到子细胞中。
现在我们知道,生物的性状(形态特征和生理生化特性)由基因决定,受环境影响,基因是染色体上具有遗传效应的DNA片段。DNA片段中的遗传信息蕴含在DNA链上A、T、C、G四种碱基的排列顺序中。
我们还知道,基因的表达遵从中心法则,从DNA转录得到mRNA,mRNA翻译得到蛋白质,蛋白质直接体现性状。转录遵循碱基互补配对原则,翻译时三个碱基是一个密码子,决定一个氨基酸。
那么,生物表现出的种种性状,最终还是由染色体上的DNA序列决定的。因此,DNA序列中的一些变化(插入、缺失、替换等)就可能造成表型的变化,引发代谢障碍,甚至引发疾病。
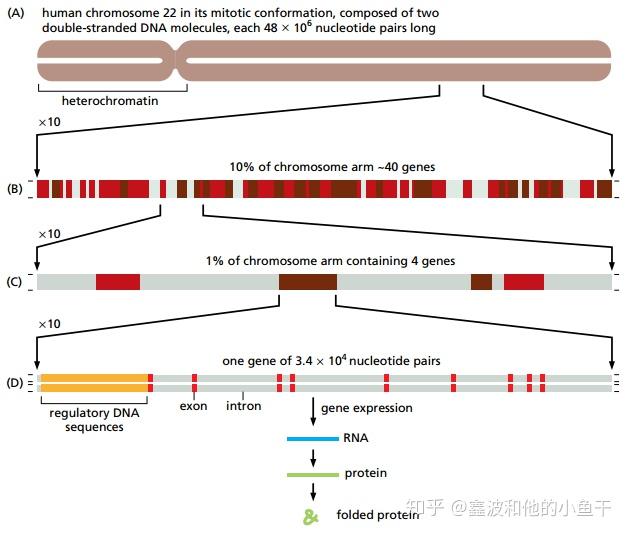
(A),人类22号染色体包含48 x 10^6对核苷酸,约占人类基因组的1.5%。(B),22号染色体的一部分,大约有40个基因。深棕色的是已知基因,红色的是预测基因,灰色是基因间序列。(C),B中显示四个基因的扩大部分。(D),典型基因的内含子-外显子排列结构,黄色是表达调控区域,红色是外显子,灰色是内含子。
<hr/>了解了上述背景,我们就可以开始讨论基因编辑啦,先说基因编辑是什么。
官方一点的说法,基因编辑,就是对目标基因及其转录产物进行编辑(定向改造),实现特定DNA片段的加入、删除,特定DNA碱基的缺失、替换等,以改变目的基因或调控元件的序列、表达量或功能。基本原理是通过序列特异性的DNA结合结构域和非特异性的DNA修饰结构域组合而成的序列特异性核酸内切酶,识别染色体上的DNA靶位点,进行切割并产生DNA双链断裂,诱导DNA的损伤修复,从而实现对指定基因组的定向编辑。
简单来说,基因编辑就是利用一个蛋白或融合蛋白作DNA分子的手术刀对目的基因进行改造。该蛋白的一部分结构可以识别/结合你要编辑DNA的特定区域(这样才能把手术刀带到需要改造的基因那里),一部分结构可以对DNA进行操作(发挥手术刀的功能,定点定向改造)。
因此,为了实现特定基因的编辑,首先要在该基因内选择适合的靶点(适合被识别、切割),然后要把酶(负责切割DNA或者替换碱基)带到这个基因的靶DNA区域,之后酶就能引起双链断裂或碱基替换等……
<hr/>这里,我们再介绍几种包括CRISPR在内的技术手段,讨论如何实现基因编辑。
1. Cre-lox 介导的基因组特定位点重组
Cre-lox 重组技术源自大肠杆菌的P1噬菌体,目前被广泛用于特异位点的基因敲除、插入、翻转和基因易位,从基因水平对生物体进行定向遗传改造,特别是在小鼠转基因中,能提供复杂的基因表达时空控制。该系统由两个来自P1噬菌体的成分组成:Cre重组酶和loxP识别位点。
Cre重组酶特异性识别 loxP位点,并负责分子内和分子间的重组,只要基因组中含有loxP位点,在细胞中表达Cre酶就能使该位点发生DNA重组,进而实现lox位点的定点编辑。
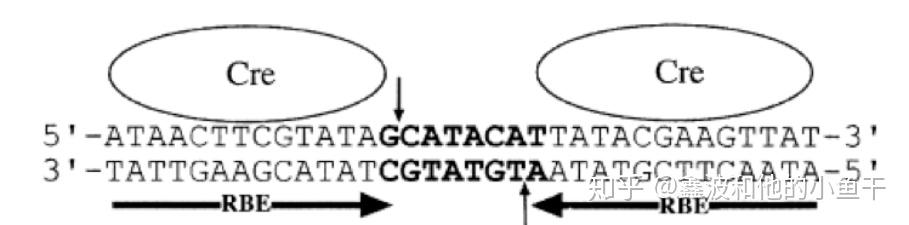
Cre酶和lox位点
Cre酶由大肠杆菌噬菌体P1的Cre基因编码,是由343个氨基酸组成的38kD的蛋白质。它不仅具有催化活性,而且与限制酶相似,能够特异性识别loxP位点。Cre酶可以单独作用,独立于其他辅助蛋白或辅助因子。
LoxP 是34对的碱基识别序列,由两个13 bp长的回文重复序列组成,中间由8 bp非对称间隔序列分隔。核心序列中的不对称使loxP站点具有方向性,除了P1噬菌体外,loxP序列在任何其他已知基因组中都不是自然发生的,而且它的长度足够长,几乎不可能随机发生。
因此,如果我们人为在基因组加入loxP位点后,一旦有Cre重组酶,便会结合到loxP位点两端的反向重复序列区形成二聚体。此二聚体与其他loxP位点的二聚体结合,进而形成四聚体。随后,loxP位点之间的DNA被Cre重组酶切下,切口在DNA连接酶的作用下重新连接。重组的结果取决于loxP位点的位置和方向。主要存在几下几种重组方式:
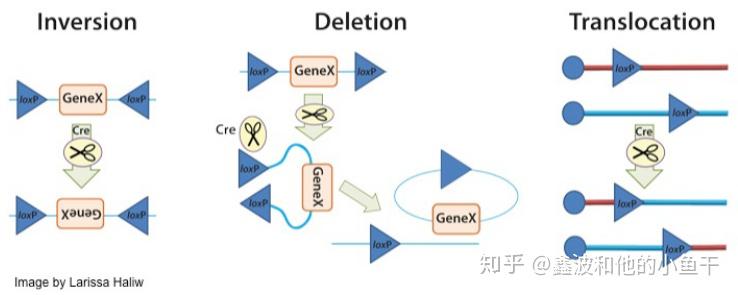
1,两个loxP位点位于同一条DNA链上且方向相同,Cre重组酶敲除loxP间的序列; 2,两个loxP位点位于同一条DNA链上且方向相反,Cre重组酶诱导loxP间的序列翻转; 3,两个loxP位点位于不同的DNA链或染色体上,Cre重组酶诱导两条DNA链发生交换或染色体易位。
该系统具有以下优点:简约高效、特异性强、应用广泛、可控的时空特异性。已成功用于功能基因的激活、转基因筛选表及的删除、基因的定点整合等。缺点是只能在固定位置插入或删除基因,不能实现对基因组中任意基因的编辑,操作受限。
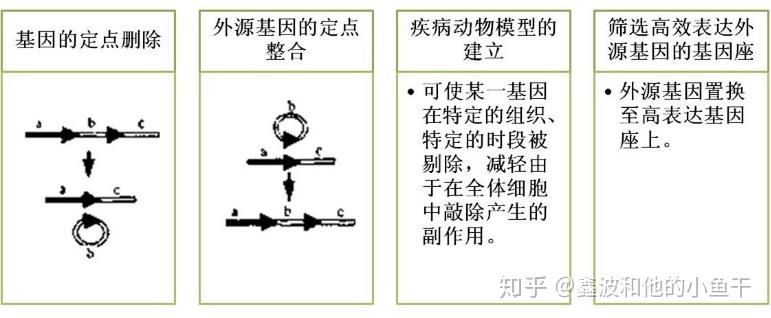
Cre-loxP重组系统在转基因中的应用
2. 锌指核酸内切酶 Zinc figer nucleases 定向修饰靶基因
锌指核酸内切酶 (ZFNs) 是人工设计的含有两个功能结构域的蛋白核酸内切酶,包括 Fok I 切割结构域 和 重复锌指结构组成的DNA识别结构域。每一个锌指结构可以特异地识别 3个核酸,6 个锌指结构就可以特异地识别 18 个核酸,从而决定了识别位点的特异性。核酸内切酶 Fok I 必须形成二聚体才能够将 DNA 双链剪开,因此要在目的位点左右两端都设计锌指酶。
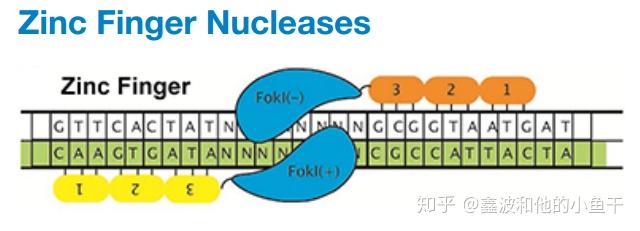
锌指核酸内切酶造成双链DNA断裂示意图
锌指核酸内切酶 将DNA双链剪开形成双链缺口,修复该缺口有两种方式:可能造成DNA缺失/插入的易错非同源末端连接 或 保守的同源修复。非同源末端连接容易形成移码突变,过早的形成终止密码子,从而造成基因敲除。保守的同源修复常规状态下会将缺口恢复如初,如果人为转入两端连有缺口同源臂的外源基因,那么就有机会将该外源基因插入到缺口中,形成基因敲入。
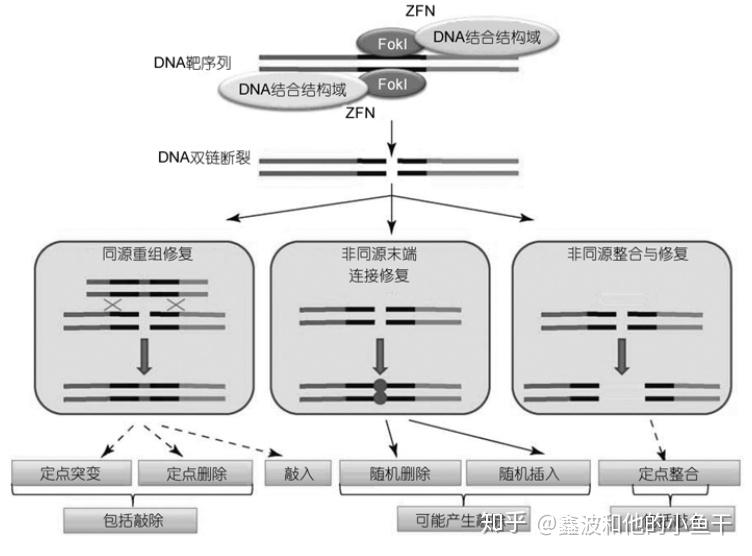
锌指核酸内切酶介导的基因组修饰途径
锌指酶基因修饰具有以下优点: 定向高效,定点精确,可以在基因组范围内实现定点敲入/敲除。缺点是锌指酶识别的目标序列长度是一定的,所以某些基因包括一些小基因和同源性高的基因,不可以被敲除,大片段基因难以通过锌指酶技术敲入。此外,锌指酶具有一定的潜在脱靶效应 。另外,由于ZFN的合成时间长,装配过程非模块化,其实用性也受到了限制。
3. 类转录激活因子效应物核酸酶 (transcription activator-like effector nuclease) 定向修饰靶基因
类转录激活因子效应物核酸酶 (TALENs) 由来自于植物病原菌黄单孢菌的类转录激活因子效应因子 TALEs 和 核酸内切酶 Fok I 的催化区域融合而成。高度保守的33~35个氨基酸TALEs重复组件决定了TALEs结合DNA的识别特异性。TALENs 性质与锌指核酸内切酶相似,识别特异的DNA序列,将其剪切形成双链缺口,通过同源修复或非同源末端连接修复,造成了基因插入或缺失。
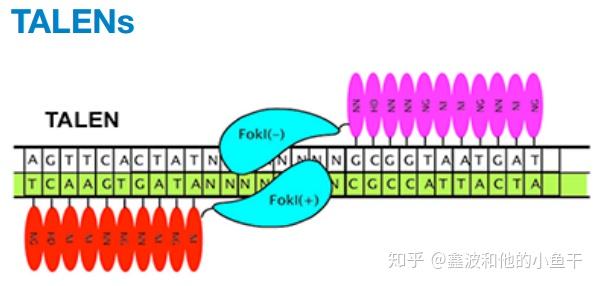
在 TALEs 中,被 称 为 repeat variable di-residues( RVD) 的 12 和 13 位相邻的两个氨基酸决定了识别的位点 。天冬酰胺和异亮氨酸识别 A,天冬氨酸和组氨酸识别 C,天冬酰胺和甘氨酸酸识别 T,两个天冬酰胺识别 G 。
TALENs 于2011年首次报道,代表了基因组工程的巨大进步。 一位经验丰富的科学家六周才能制作ZFN,但新手可以在短短几天内制作一个TALEN!TALENs 的可定制DNA结合特性还使得定制转录因子的设计能够调节基因表达。TALENs 和 ZFN 相比,操作简便,而且TALENs 的识别位点更广泛,几乎可以敲除所有的基因,TALENs 的剪切效率比ZFN高。
4. 如日中天的 CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 系统
CRISPR 近年来很火,大家也都比较熟悉了,它的中文翻译很绕口,叫“成簇排列的有规律间隔的短回文重复序列”,CRISPR/Cas系统作为细菌的获得性免疫,用于抵御噬菌体入侵。
简单来说,Cas9就像限制性核酸内切酶一样,能够切割DNA。不同的是,绝大多数限制性核酸内切酶识别固定的DNA序列,而Cas9对DNA的识别需要一个sgRNA与靶DNA配对,切割需要靶DNA上的PAM序列。crRNA跟哪个DNA配对,就能把Cas9领到那里,Cas9识别PAM并对DNA进行切割。当然,也可以将Cas9的切割结构域突变掉,融合表达胞嘧啶脱氨酶之类的碱基改造酶类,就可以实现碱基的定点替换。
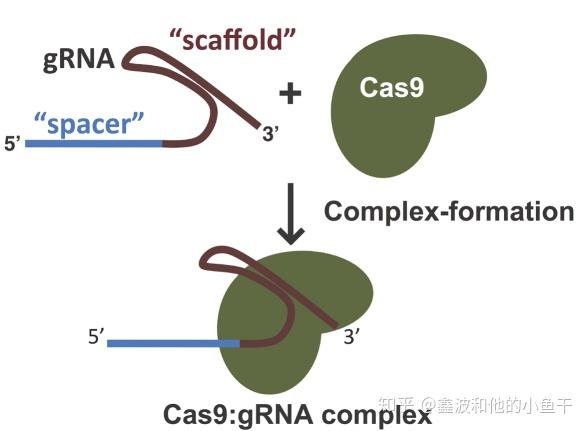
与靶DNA配对的是spacer(外来DNA上截下来的记忆片段)转录并加工出的crRNA,tracerRNA作支架与crRNA一起结合Cas9,对靶DNA进行切割。为了操作方便,提高效率,科学家们把crRNA和tracerRNA融合到一起,称作single guide RNA,即sgRNA。
因此,如果我想编辑一个基因,首先要选择这个基因上的PAM位点附近作靶点,然后设计sgRNA,在细胞表达sgRNA和Cas9,就可以将目的片段剪切形成双链缺口,通过同源修复或非同源末端连接修复,造成了基因插入或缺失。如果同时转入有同源臂的外源基因,那么就有机会将该外源基因插入到缺口中,形成基因敲入。
CRISPR技术的优点是,能够实现定点编辑,可一次性敲除多个基因,适用对象广泛,使用简单,成本低。缺点是切割受靶DNA上的PAM限制,偶有脱靶。
考虑到CRISPR的应用越来越广泛,其原理和操作也相对简单,这里我们就展开介绍一下,不想深入了解的小伙伴可以跳过,直接看小结。
细菌通过以下两步描述 CRISPR 发挥二次免疫:
(1) 获得 spacer 记忆,在 Cas1 和 Cas2 及 Csn2 蛋白的作用下, 细菌识别入侵核酸并扫描外源DNA潜在的PAM,将临近 PAM 的序列作为候选spacer,然后在CRISPR基因座的 5&#39;端合成repeats;最后新的spacer整合到两个repeat之间,CRISPR基因座中的spacer从5&#39;到3&#39;的排列记录了外源遗传物质入侵的时间顺序。
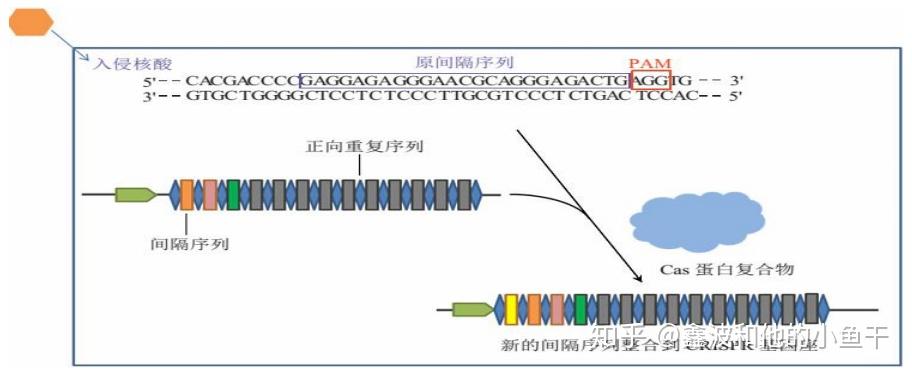
初次侵染后,拥有CRISPR/Cas系统的细菌会截取噬菌体DNA的一部分作为间隔序列,储存在自己基因组上规律存在的重复序列之间,作为记忆。细菌再次被同种噬菌体侵染时,之前保留的记忆(截下来的DNA)会转录出crRNA,这个RNA可以与tracerRNA一起结合Cas9,去配对并靶向降解外来的噬菌体DNA,预防侵染。
(2) 发挥免疫功能,发挥免疫的过程可以再划分为两步:① CRISPR表达盒内基因的表达,CRISPR基因座首先被转录成前体CRISPR RNA(pre-crRNA),在Cas蛋白或核酸内切酶的作用下被剪切成一些成熟的小RNA单元称crRNA,TypeⅡ型 CRISPR/Cas系统还需要tracrRNA的指导。②基因表达产物组装,切断外源DNA。成熟的crRNA与特异的Cas蛋白形成复合物,再与外源DNA结合并扫描到外源DNA,寻找其上的靶序列,crRNA的spacer与靶序列互补配对,外源 DNA在配对的特定位置被核糖核蛋白复合物切割。
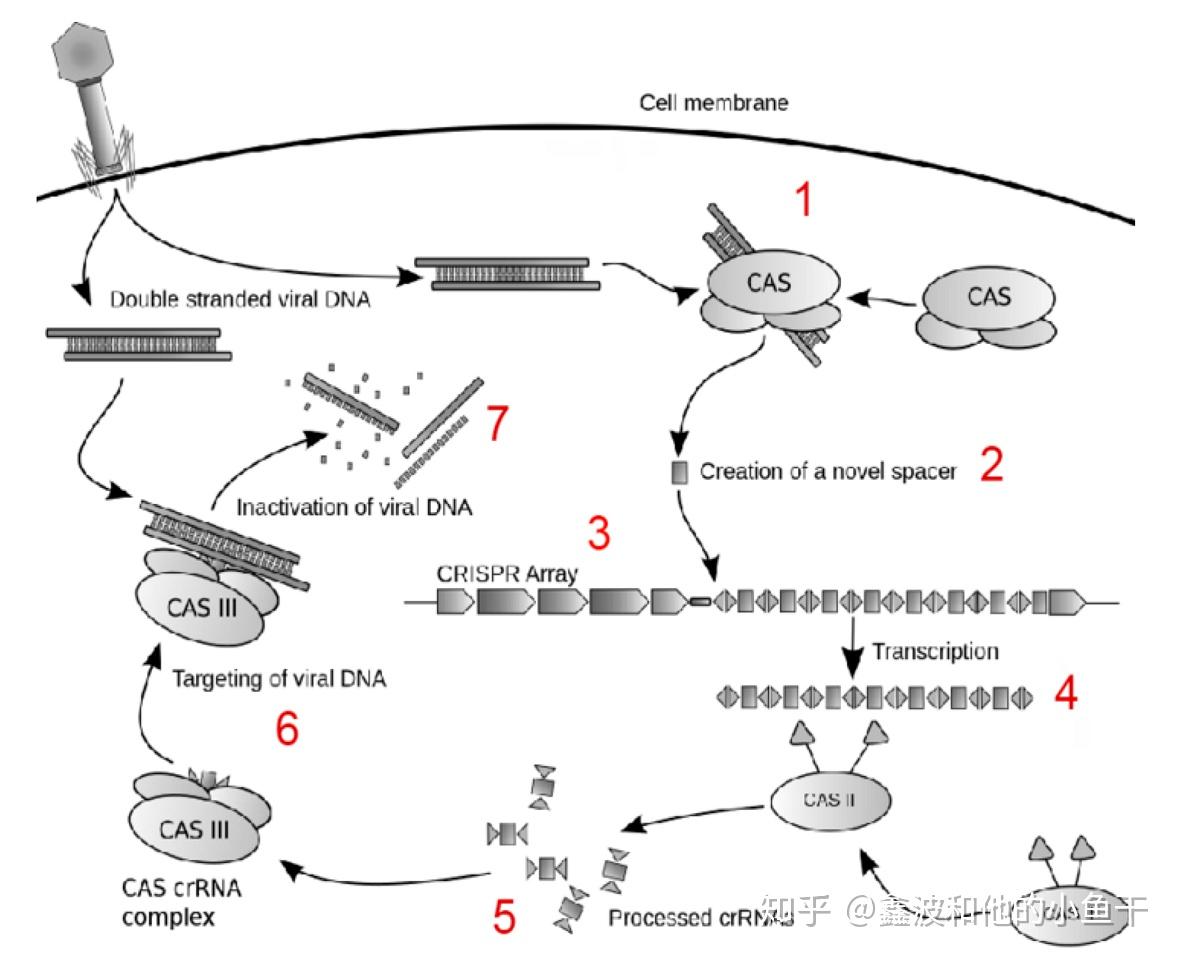
类似于人的免疫反应,CRISPR免疫也有初次免疫和二次免疫之分(虽然大家不这么说) :初次免疫即噬菌体DNA侵入,细菌对外源基因截取作为spacer保存在repeats中;再次免疫即细菌的基因组中已经含有了该噬菌体对应的spacer,CRISPR系统快速降解入侵DNA的过程。
在CRISPR/Cas系统的作用机制被揭示清楚后,科学家们认识到其可能具有巨大的应用前景。基于以前ZFN及TALEN编辑技术的应用范围,人们推测 CRISPR/Cas系统也可能在其他的真核生物中工作。基于第二类 CRISPR,人们开发了 CRISPR/Cas9 系统用于基因编辑,实验结果证明将 tracerRNA 与 crRNA 用一个小片段连接成为 sgRNA 后,不影响该系统正常工作且打靶效率有所提高,后来常规的表达策略就将 sgRNA 与 Cas9 整合到一个表达载体上。
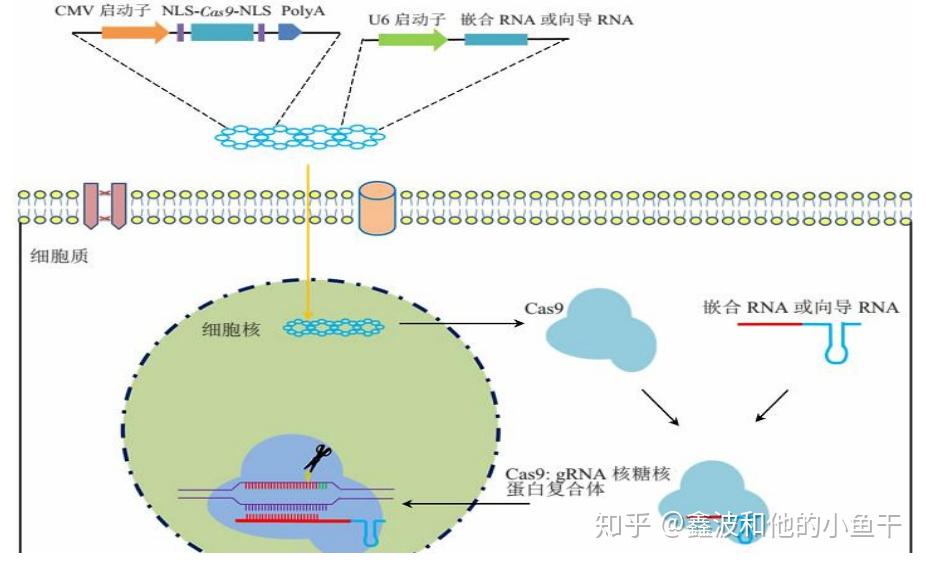
CRISPR/Cas9 用于真核生物基因敲除示意图。表达的Cas9蛋白在核定位信号的引导下进入细胞核,Cas9结合sgRNA后识别染色体上特定DNA区段,由Cas9的切割结构域造成DNA双链断裂。
CRISPR/Cas9 系统在真核生物中的打靶效率受到靶位点染色体状态的影响,要让 Cas9 蛋白高效地转运到哺乳动物细胞核内,需在 Cas9 蛋白的N端或C端加上真核细胞的核定位信号(NLS)。Cas9 蛋白在折叠过程中对临近的NLS功能有一定“遮蔽”作用,N 端核定位信号和Cas9 蛋白之间要加上32个氨基酸残基的接头才能发挥功能,在 Cas9 蛋白的C端添加核定位信号似乎更有效。
关于CRISPR的一些问题
1,细菌天然 CRISPR 如何避免自身免疫 成熟的crRNA除spacer外,其5&#39;端和3&#39;端含有部分repeat序列,当外源DNA入侵时,crRNA 两端序列不能与靶位点配对,对于自己CRISPR盒子中的相同序列,crRNA与两端的repeat序列也能完全配对,实际上只有不完全的配对才允许核糖核蛋白复合体发挥切割活性。与细菌整合进CRISPR盒的spacer对应的噬菌体片段被称为protospacer,其5&#39;或是 3&#39;端相隔1-4bp,有一个长为2-5bp的相对保守序列,该序列被称为PAM。在细菌CRISPR盒子里的spacer没有PAM,这样CRISPR发挥免疫的时候就不会切掉自己,也就避免了细菌的自身免疫。
2,免疫的有效性, 并不是每种细菌只含一类 CRISPR 系统,也有些同时拥有两类或三类,这是细菌与病毒共同进化的结果,但这并不说明CRISPR总是有效的。噬菌体的基因也有变异,很多发生了点突变的噬菌体DNA就无法被先前的记忆(spacer)所识别,这就发生了“免疫逃逸” 。
3,靶 DNA 怎么被切开的 CRISPR/Cas 系统的作用特性与限制性核酸内切酶相似,切割特异性主要依赖于识别靶序列上的PAM以及protospacer。Cas9蛋白存在HNH和RuvC两个活性位点,可分别切割与crRNA互补的DNA链和非互补链,切割位点位于PAM上游3nt处,RuvC-like结构域可以对另一条链进行切割,切点于PAM上游3-8nt处,切割后形成双链断裂(DSBs)。
4,切开以后的 DNA 会怎样 DNA损伤后产生的DSBs激活细胞内固有的损伤修复。不存在修复模板时,NHEJ用于双链断裂的连接,造成插入/缺失(indel)突变;HDR通过模板进行同源修复,可将序列复制到双链断裂处,将特定的核苷酸或片段引入目标基因。
5,脱靶 前人发现最多5个碱基存在错配情况时,系统仍会对该位点进行切割,造成脱靶。研究表明,CRISPR/Cas 系统在细菌中是特异的,在水稻中特异性也很高。而 CRISPR/Cpf1 系统应用于基因组精确编辑几乎不存在脱靶效应。
<hr/>划重点
Cre-lox系统中,cre酶特异性识别lox位点,将“手术刀”带到特定DNA区域,通过cre酶的DNA重组活性进行操作。
锌指核酸内切酶 (ZFNs) 系统中,串联的锌指结构识别基因组中对应的DNA位点,想操作哪里的DNA,就根据那里的DNA序列设计串联锌指,将“手术刀”带到特定DNA区域。锌指核酸内切酶 (ZFNs) 通过Fox I 的DNA内切酶活性进行操作。
类转录激活因子效应物核酸酶 (TALENs) 系统中,类转录激活因子效应因子识别基因组中对应的DNA位点,将“手术刀”带到特定DNA区域,而后通过Fox I 的DNA内切酶活性进行操作。TALENs 比 ZFN 设计简便,识别位点广,剪切效率高。
CRISPR/Cas系统中,sgRNA配对识别基因组中对应的DNA位点,将“手术刀”带到特定DNA区域,而后Cas9识别该区域附近DNA上的PAM,通过Cas9的DNA内切酶活性切割DNA。
所有这些编辑技术用于敲除时,都只是切割DNA,产生双链断裂。编辑结果是细胞对DNA双链断裂的错误修复造成的。
<hr/>如何对待基因编辑
对于基因编辑用于动植物育种,我一直都是很支持的。它跟转基因也有本质区别,肯定会更早投入生产应用。
对动植物的基因编辑,最终产生的是对该物种自身基因的改变,基于自身基因的定向改造不涉及外来基因,这就和自然状态下的变异没有区别,产生的大片段缺失和X射线诱变育种的缺失没有区别,甚至因为更加精准而更安全可靠。
对于很多植物科学的研究成果,可以通过基因编辑,敲除、偏低或者碱基定向替换就可以快速投入生产的,如果基因编辑受到太多监管、约束和限制,这些研究成果的推广就会比较受限。因为自然状态下突变的低频性,不容易获得目标基因的突变,如果不同生态型该基因没什么变化,那就只能定向创造突变才能推广该成果。
比如,OsLCT1编码了一个负责将重金属Cd镉转向水稻籽粒的转运蛋白,我们敲除它就能降低水稻籽粒重金属含量。但是不同品种、亚种中该基因的自然变异差别极小,我们不能通过传统杂交、回交来实现该目标,只能通过基因编辑实现。如果限制基因编辑在作物育种中的应用,那么我们科研中的种种发现就很难推广到实际生产应用了。
<hr/>课后思考题
- 本文所述的四种基因编辑工具原理分别是什么,谁识别DNA,又是谁切割DNA?
2. 如果我想使用CRISPR技术,对一个目的基因进行敲除,我需要设计什么,注意什么?
参考文献
陈广东, 崔继哲, 付寅生, et al. Cre/lox位点特异重组系统在植物基因工程中的应用[J]. 生物技术通报, 2012(5):25-30.
何祖勇, 梅瑰, 赵春鹏, et al. 基于FoldX力场的蛋白突变建模方法可用于人工锌指酶辅助设计[J]. 中国科学:生命科学, 2011(2):121-128.
王贵利, 张连峰. 基因工程大鼠研究进展[J]. 中国比较医学杂志, 2013, 23(3):71-76.
方锐, 畅飞, 孙照霖, et al. CRISPR/Cas9介导的基因组定点编辑技术[J]. 生物化学与生物物理进展, 2013, 40(8):691-702.
科学网-Cre-loxP重组系统介绍
CRISPR三类系统的详细解读
基因敲入相比于基因敲除有哪些难点? |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-16 18:22
发表于 2024-9-16 18:22







