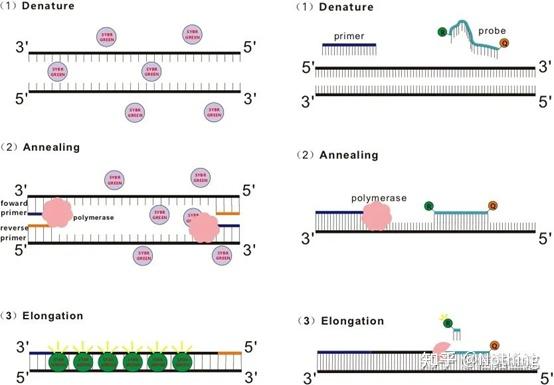
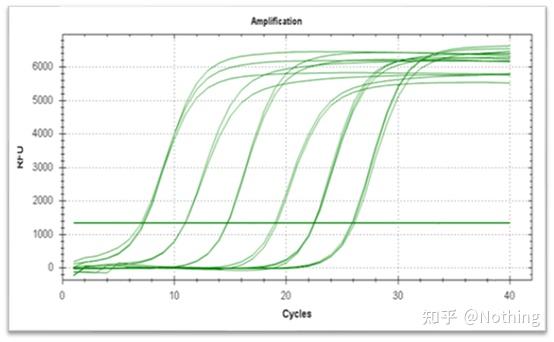
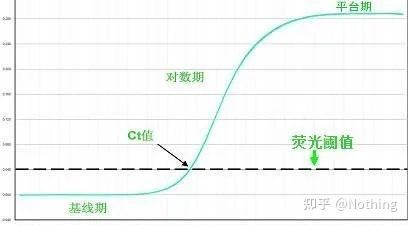
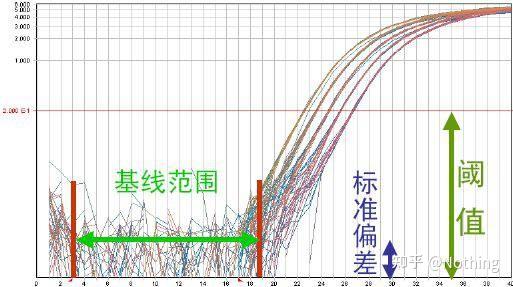
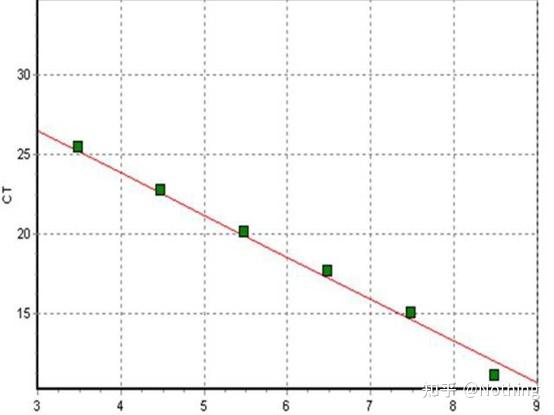
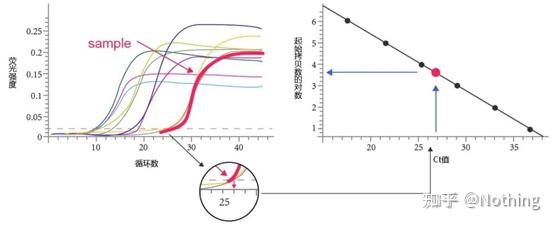
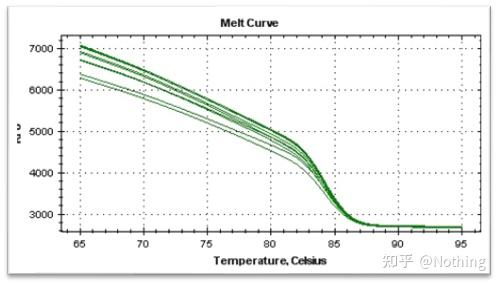
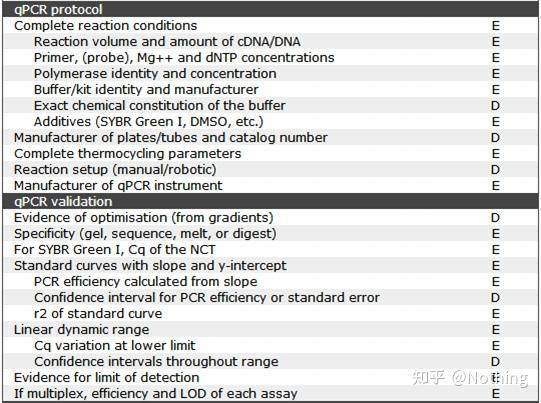
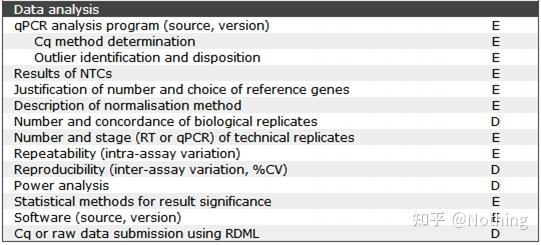
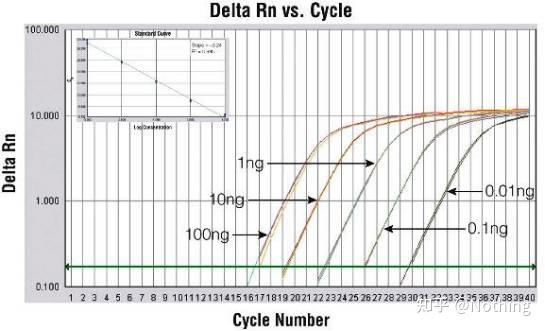
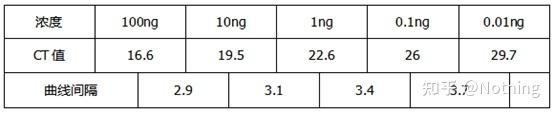
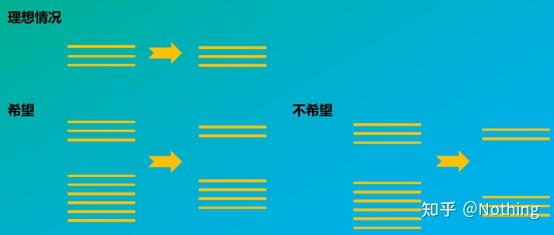
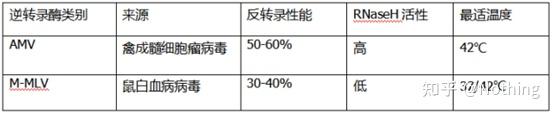
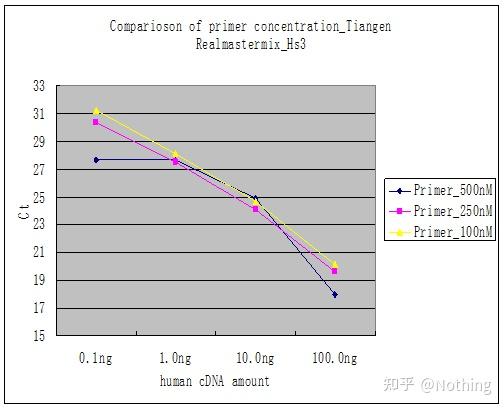
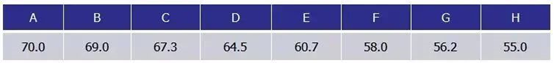
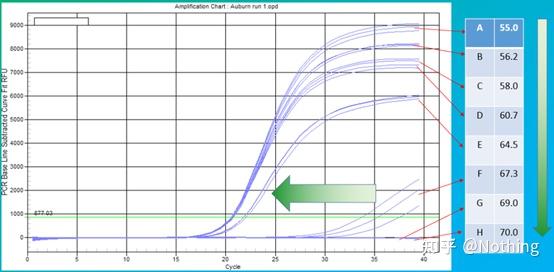
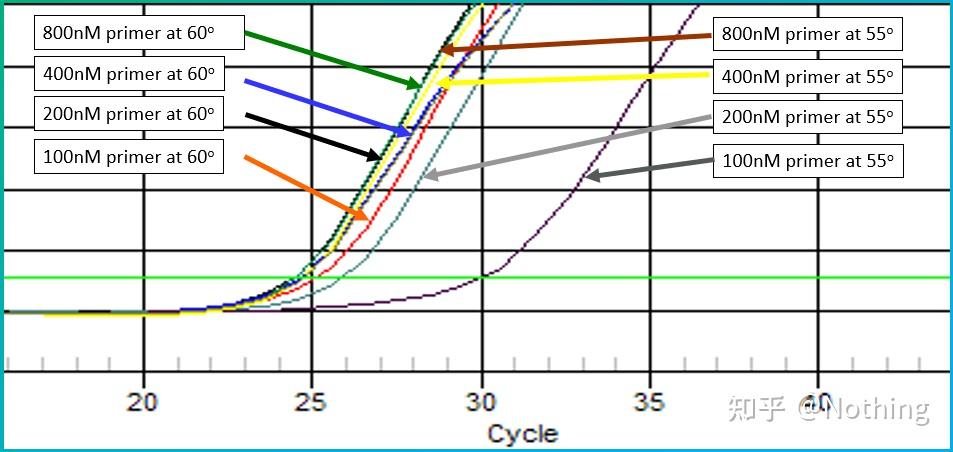
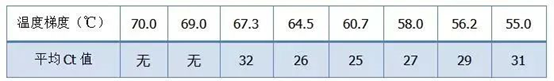
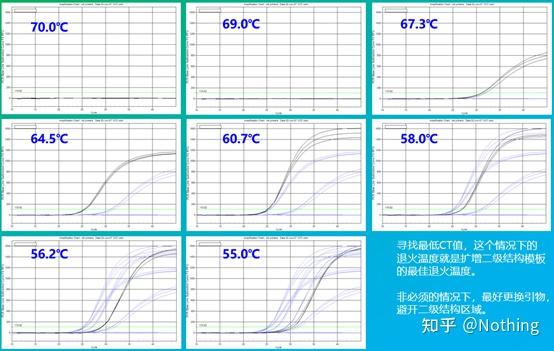
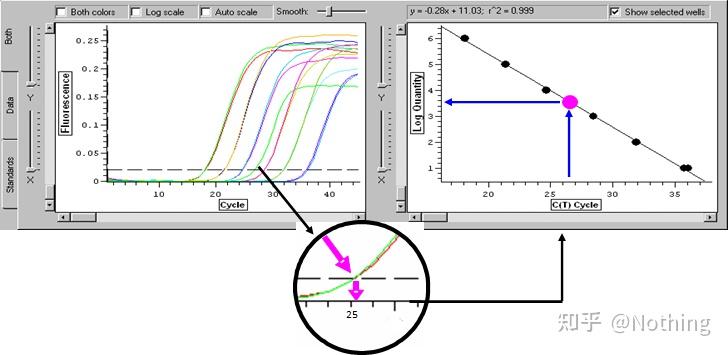
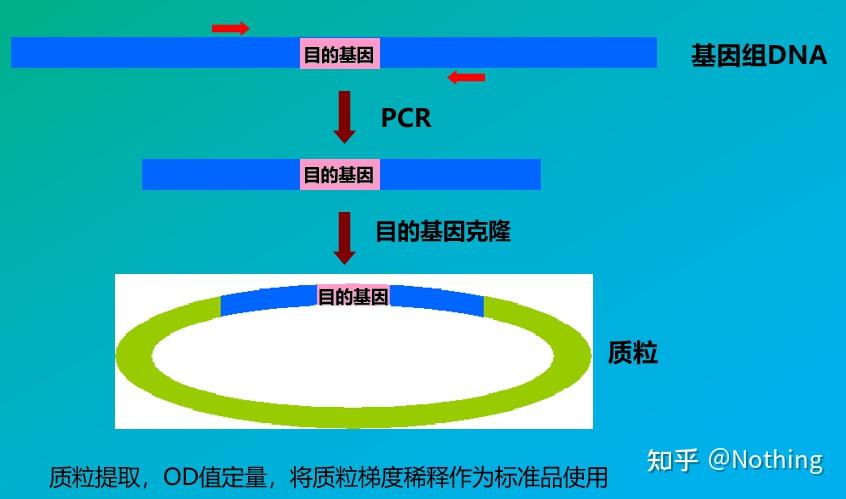
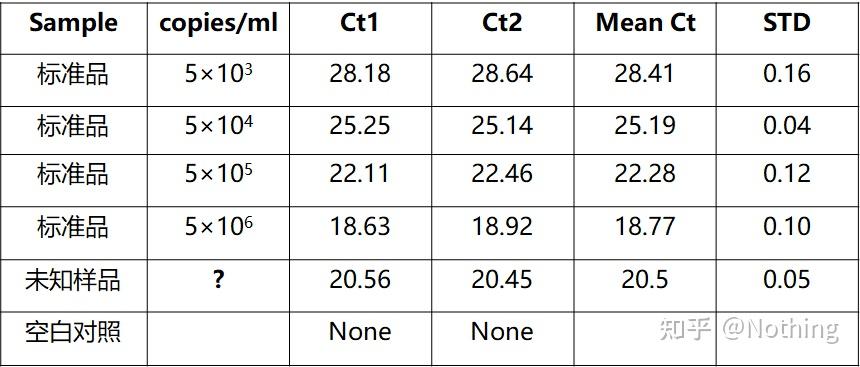
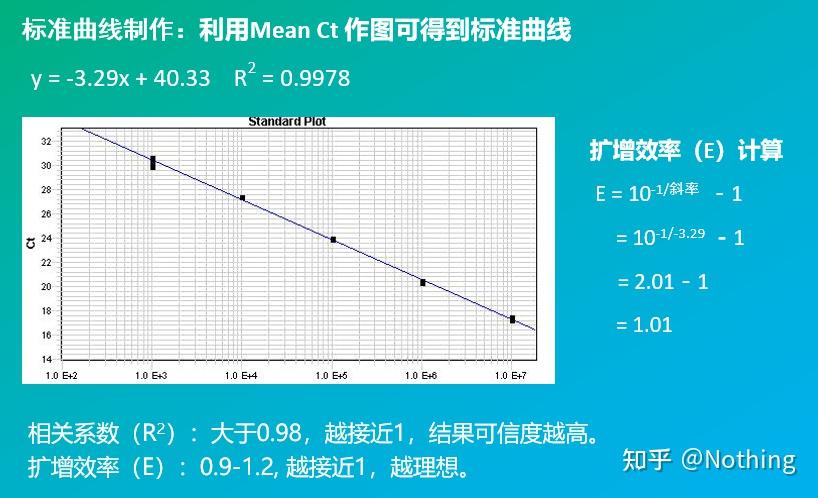
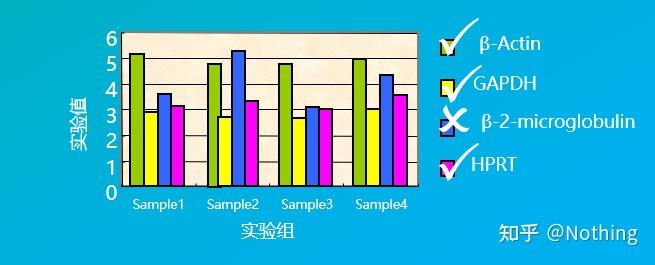
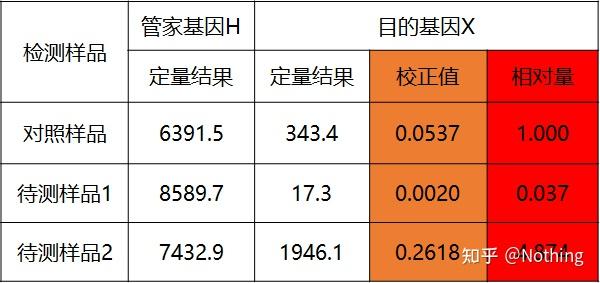
答:染料法 一些荧光染料如SYBR Green Ⅰ,PicoGreen,BEBO等,它们本身不发光,但结合于双链DNA的小沟后会发出荧光。所以当PCR反应刚开始时机器并不能检测到荧光信号,当反应进行到退火-延伸阶段,此时双链打开,在DNA聚合酶的作用下新链合成,荧光分子就结合于dsDNA的小沟中并发出荧光,随着PCR循环数的增加越来越多的染料与双链DNA结合,荧光信号也不断的增强。染料法主要应用于科研。
PS:做实验小心点,染料可是要跟人的DNA结合的,小心变成荧光人。
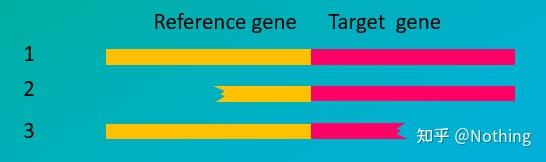
染料法(左)探针法(右)
PS:做实验小心点,染料可是要跟人的DNA结合的,小心变成荧光人。
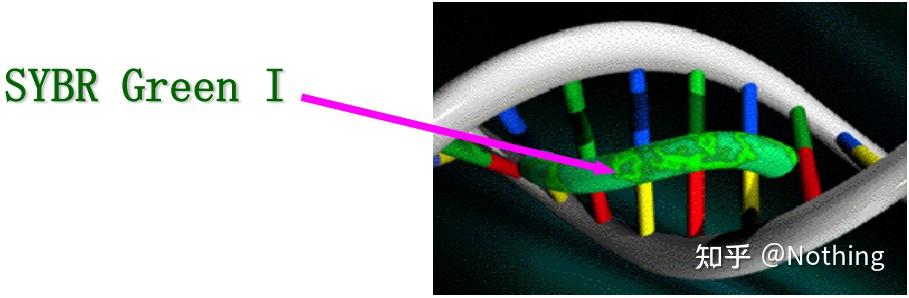
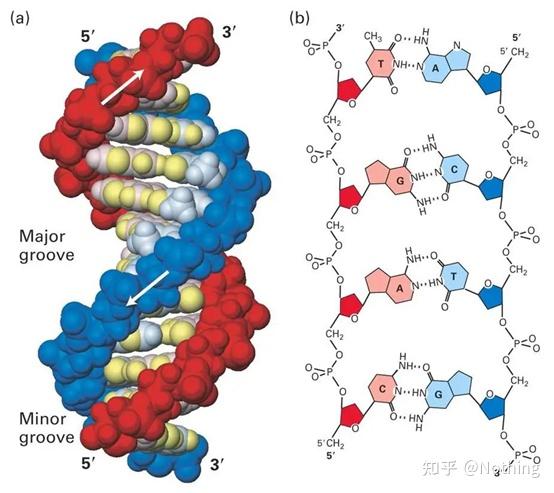
SYBR GreenⅠ与DNA小沟结合
探针法 Taqman探针是最为常用的一种水解探针,在探针的5’端存在一个荧光基团,通常为FAM,探针本身则为一段与目的基因互补的序列,在探针的3’端有一个荧光猝灭基团,根据荧光共振能量转移原理(Förster resonance energy transfer, FRET),当报告荧光基团(供体荧光分子)和猝灭荧光基团(受体荧光分子)激发光谱重叠且距离很近时(7-10nm),供体分子的激发可以诱发受体分子发荧光,而自身荧光减弱。所以PCR反应开始,探针游离于体系中完整存在时,报告荧光基团并不会发出荧光,当退火时,引物和探针结合于模板,在延伸阶段,聚合酶不断的合成新链,由于DNA聚合酶具有5’-3’核酸外切酶活性,到达探针时,DNA聚合酶就会将探针从模板上水解下来,报告荧光基团和猝灭荧光基团分开,释放荧光信号。由于探针和模板存在一对一的关系,所以在试验的精度和灵敏度上,探针法都要优于染料法。探针法主要应用于诊断。
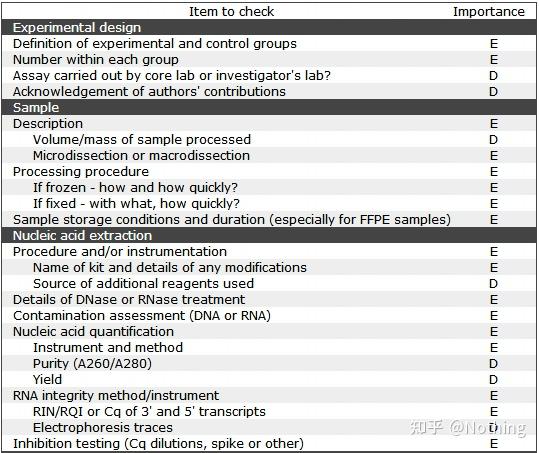
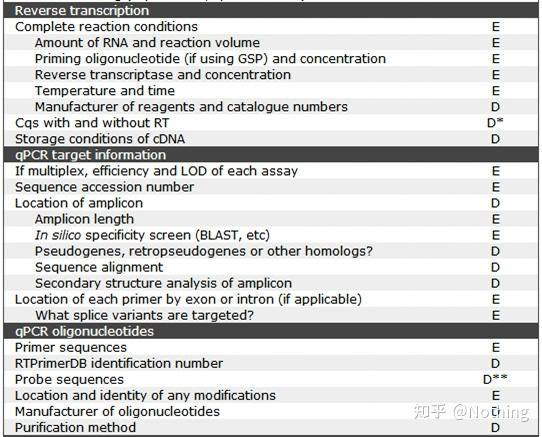
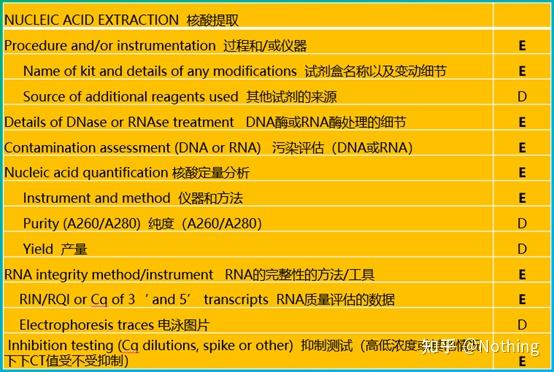
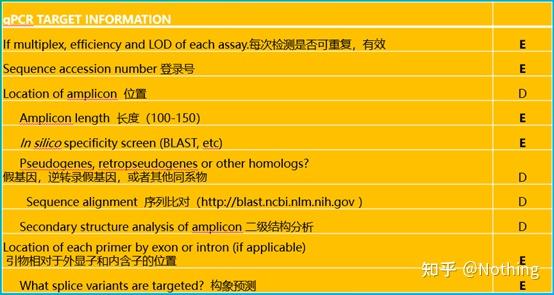
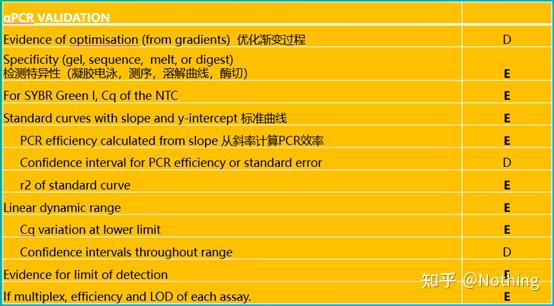
MIBBI(Minimum Information for Biological and Biomedical Investigations - http://www.mibbi.org) 应运而生。MIBBI是一个为实验提供标准的项目,发表在nature上,这个项目针对各类生物学试验,包括细胞生物学、Microarray、我们现在要讨论的qPCR等等,给每一类实验规定了在提交稿件时是都应该提供那些信息。
MIBBI项目中,与荧光定量PCR相关的文章有两篇,分别是:
RDML (Real-Time PCR Data Markup Language )——实时定量PCR数据的结构化语言和报告指南;
MIQE(Minimum Information for Publication of Quantitative Real-Time PCR Experiments)——用于发表关于实时定量PCR实验文章的最小信息。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-13 07:22
发表于 2024-9-13 07:22