金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
关注公众号“医学科研小坑”,了解更多相关科研技术内容!
免疫印迹法 (Western blotting) 是一种将高分辨率凝胶电泳和免疫化学分析技术相结合的杂交技术。通俗来讲,将蛋白质通过电转固相到膜上,然后利用抗体检测蛋白表达的方法。与Southern早先建立的检测核酸的印迹方法 Southern blot 相类似,亦被称为Western blot。
Western Blotting可用于:
1.从细胞或组织总蛋白中检测目标蛋白;
2.定量或定性确定目标蛋白表达水平;
3.蛋白-蛋白,DNA-蛋白,RNA-蛋白相互作用的检测分析。
实验原理:
蛋白研究样品通过恒定电流电泳经过PAGE胶,由于各蛋白所带分子量不同而产生不同的电泳速度而被分离。通过电转移固相到PVDF膜或NC膜,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技术也广泛应用于检测蛋白水平的表达。
科研中的运用:
(1) 检测细胞中目的蛋白的表达
通过WB技术直接检测干细胞分化、细胞自噬、细胞凋亡、周期蛋白、DNA损伤修复、分子诊断标志物等关键通路蛋白,可评估细胞或机体所处的生物学状态。
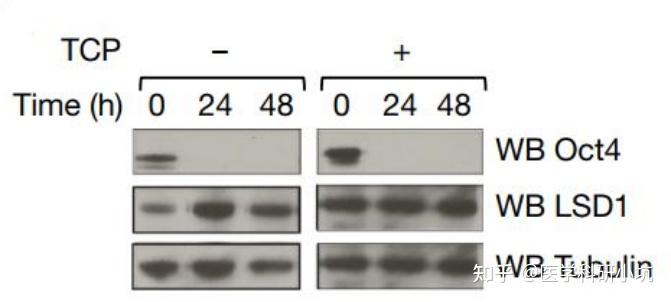
图1.通过WB检测经典Yamanaka因子评估胚胎干细胞的分化[1]
(2) 对基因的功能缺失研究
通过敲减或过表达目的基因后,用WB技术检测相关通路蛋白的表达变化,以确定目的基因激活或抑制目的蛋白的表达调控关系,探究信号通路。
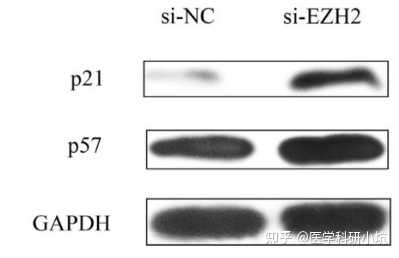
图2. WB证明敲减EZH2上调p21蛋白表达[2]
(3) 研究目的蛋白所处的细胞定位
蛋白的分选投递与发挥功能是基因调控的结果,因此改变了某些基因的活性,或化学修饰可能会影响其效应蛋白的转运,改变细胞核质中的分布,引发不同的生物学效应。
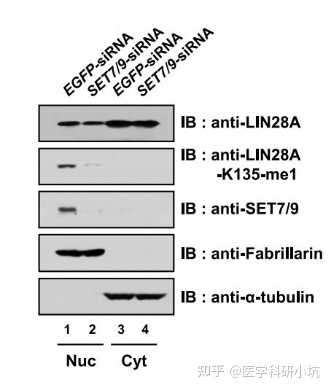
图3.通过WB检测目的蛋白LIN28A在细胞核质中的分布[3]
(4) 药物处理、代谢类的研究
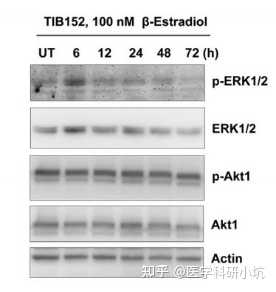
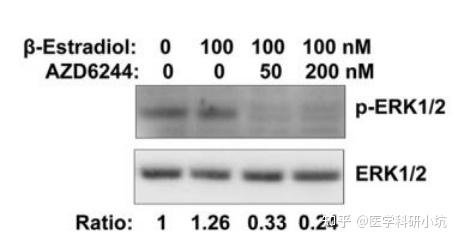
图4.研究药物处理时间或计量的不同对效应蛋白的研究[4]
(5) 蛋白相互作用类研究
通过WB可检测Co-IP、RIP、CHIRP等实验的IP产物,研究目标蛋白-蛋白,RNA-蛋白之间的相互作用。
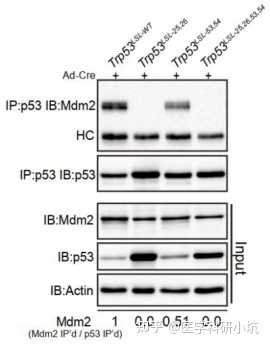
图5. 不同的Trp53等位基因不成程度的与效应蛋白MDM2相互作用[5]
实验流程:
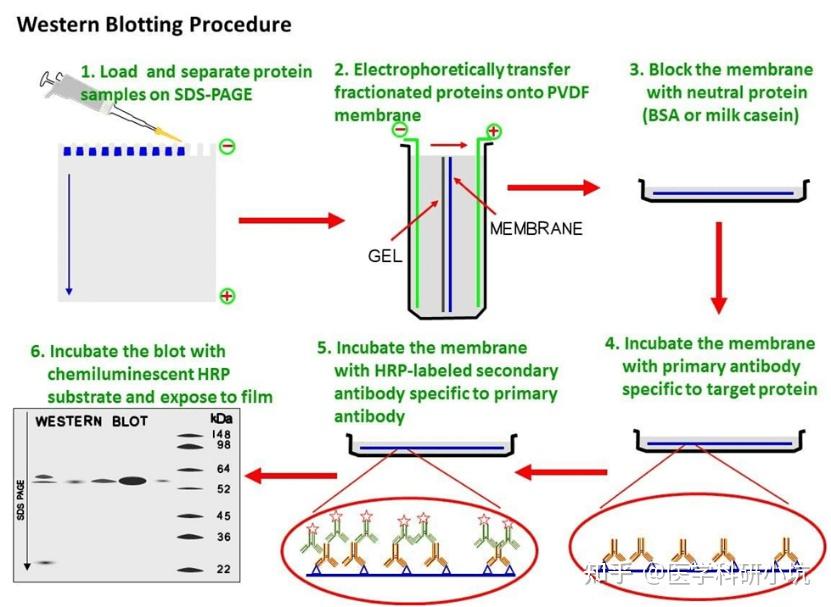
一、 试剂准备
1. SDS-PAGE试剂:见聚丙烯酰胺凝胶电泳实验;
2. 匀浆缓冲液:1.0 M Tris-HCl(pH 6.8) 1.0 ml;10%SDS 6.0 ml;β-巯基乙醇 0.2 ml;ddH2O 2.8 ml;
3. 转膜缓冲液:甘氨酸 2.9 g;Tris 5.8 g;SDS 0.37 g;甲醇200 ml;加ddH2O定容至1000 ml;
4. 0.01 M PBS(pH7.4):NaCl 8.0 g;KCl 0.2 g;Na2HPO4 1.44 g;KH2PO4 0.24 g;加ddH2O至1000 ml;
5. 膜染色液:考马斯亮兰 0.2 g;甲醇80 ml;乙酸2 ml;ddH2O118 ml。包被液(5%脱脂奶粉,现配):脱脂奶粉1.0 g 溶于20 ml的0.01 M PBS中;
6. 显色液:DAB 6.0 mg;0.01 M PBS 10.0 ml;硫酸镍胺 0.1 ml;H202 1.0 μL。
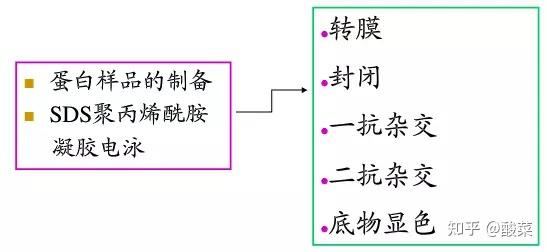
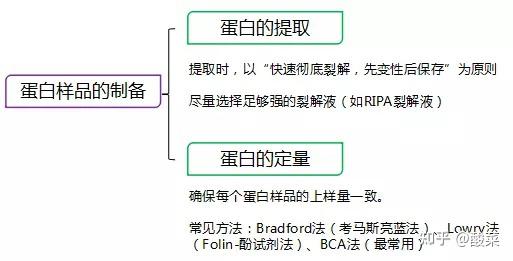
二、 蛋白样品制备
贴壁细胞样品
1. 倒掉培养液,并将瓶倒扣在吸水纸上使吸水纸吸干培养液;
2. 每瓶细胞加3 ml 4℃预冷的PBS(0.01M pH7.2~7.3)。平放轻轻摇动1 min洗涤细胞,然后弃去洗液。重复以上操作两次,共洗细胞三次以洗去培养液。将PBS弃净后把培养瓶置于冰上;
3. 按1ml裂解液加10 μL PMSF(100 mM),摇匀置于冰上;
4. 每瓶细胞加400 μL含PMSF的裂解液,于冰上裂解30 min,为使细胞充分裂解培养瓶要经常来回摇动;
5. 裂解完后,用干净的刮棒将细胞刮于培养瓶的一侧(动作要快),然后用枪将细胞碎片和裂解液移至1.5 ml离心管中;
6. 于4℃下12000 rpm离心5 min;
7. 将离心后的上清分装转移到0.5 ml的离心管中放于-20℃保存。
组织总蛋白的提取
1. 将少量组织块置于1~2 ml匀浆器中球状部位,用干净的剪刀将组织块尽量剪碎;
2. 加400 uL单去污剂裂解液裂(含PMSF或蛋白酶抑制剂)于匀浆器中,进行匀浆。然后置于冰上;
3. 几分钟后再碾一会儿再置于冰上,要重复碾几次使组织尽量碾碎;
4. 裂解30 min后,即可用移液器将裂解液移至1.5 ml离心管中,然后在4℃下12000 rpm离心5 min,取上清分装于0.5 ml离心管中并置于-20℃保存。
三、 蛋白定量
根据蛋白定量试剂盒(通常使用BCA法),对样品中总蛋白浓度进行定量。
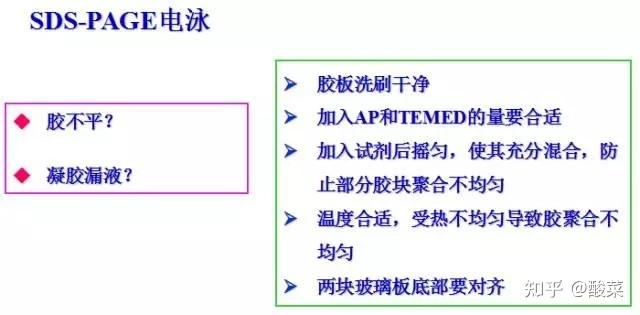
四、 SDS-PAGE电泳
1. 将样品加入等量的2×SDS上样缓冲液,100℃加热3-5min,离心12000g×1min,取上清作SDS-PAGE分析;
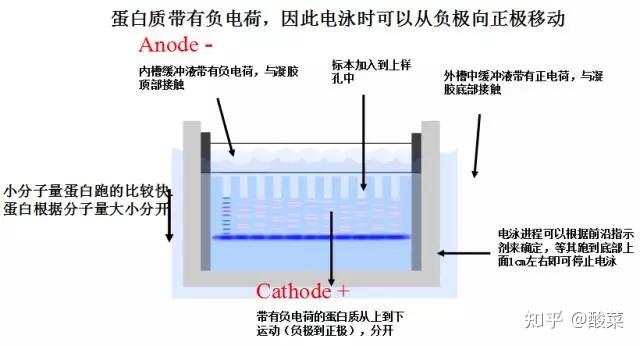
2. 电泳:在电泳槽中加入1×电泳缓冲液,连接电源,电泳时,积层胶电压80V,分离胶电压150V,电泳至溴酚兰行至电泳槽下端停止(约需2hr)。
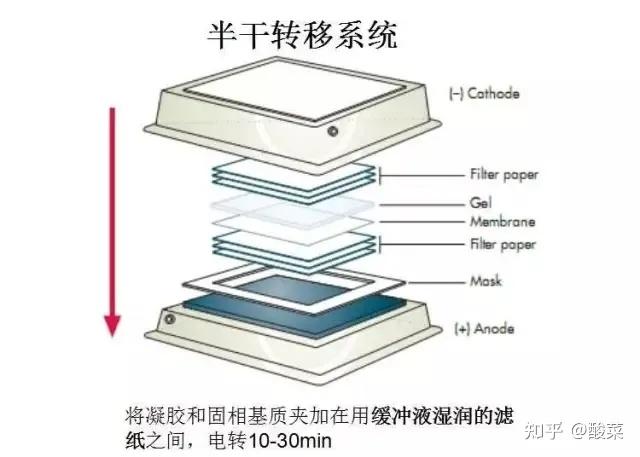
五、 转膜
1. 电泳结束后将胶条割至合适大小,用转膜缓冲液平衡,5min×3次 ;
2. 膜处理:预先裁好与胶条同样大小的6张滤纸和PVDF膜,将膜在甲醇中浸泡15秒,然后再在蒸馏水中浸泡2min,最后在转膜缓冲液中最少平衡5min;
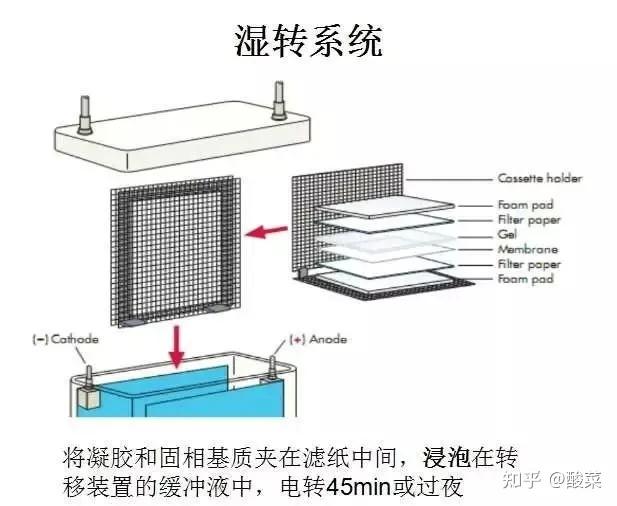
3. 转膜:转膜装置从下至上依次按阴极碳板、3层滤纸、凝胶、PVDF膜、3层滤纸、阳极碳板的顺序放好,滤纸、凝胶、PVDF膜精确对齐,每一步去除气泡。接通电源,恒流250mA,转移120min (具体转膜时间可根据目的蛋白分子量大小与转膜电流调整)。
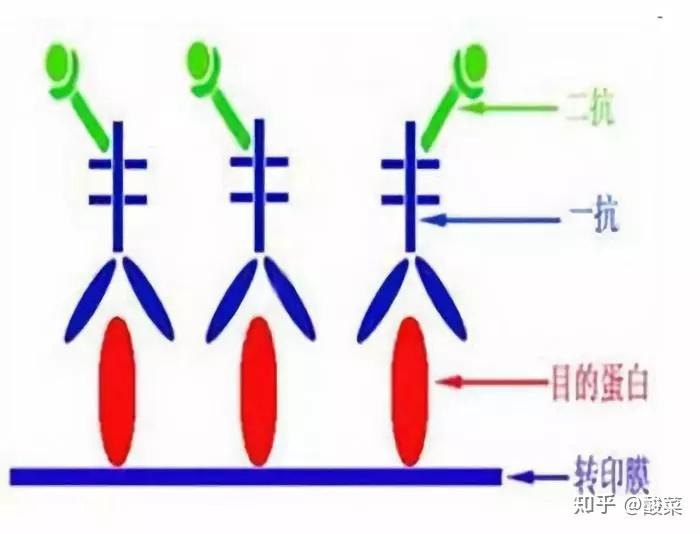
六、 免疫反应:
1. 用0.01M PBST洗膜(pH7.4, 0.02%吐 温20),5min ×3次;
2. 封闭:将纤维素膜置于一塑料袋中,加封闭液(5%脱脂牛奶),室温下孵育1小时或4℃过夜;
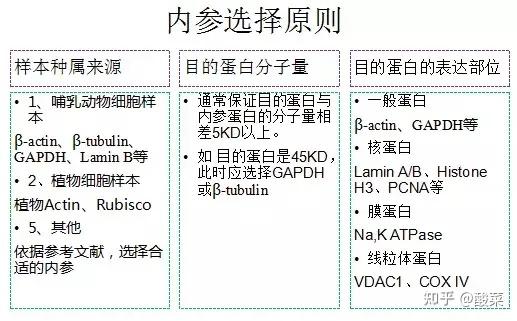
3. 加入以1mg/ml浓度,1:1000稀释的一抗抗体,室温孵育2h或4℃过夜;
4. 用TBST在室温下脱色摇床上洗两次,每次10 min;再用TBS洗一次,10 min;
5. 同上方法准备二抗稀释液并与膜接触,室温下孵育1h后,用TBST在室温下脱色摇床上洗两次,每次10 min;再用TBS洗一次,10 min,进行化学发光反应。
七、 化学发光,显影,定影
1. 将A和B两种试剂在保鲜膜上等体积混合;1 min后,将膜蛋白面朝下与此混合液充分接触;1 min后,将膜移至另一保鲜膜上,去尽残液,包好,放入X-光片夹中;
2. 在暗室中,将1×显影液和定影液分别到入塑料盘中;在红灯下取出X-光片,;打开X-光片夹,把X-光片放在膜上固定,关上X-光片夹,根据信号的强弱适当调整曝光时间;曝光完成后,将X-光片,迅速浸入显影液中显影,待出现明显条带后,即刻终止显影。显影时间一般为1~2 min(20~25℃),温度过低时(低于16℃)需适当延长显影时间;显影结束后,马上把X-光片浸入定影液中,定影时间一般为5~10 min,以胶片透明为止;用自来水冲去残留的定影液后,室温下晾干。
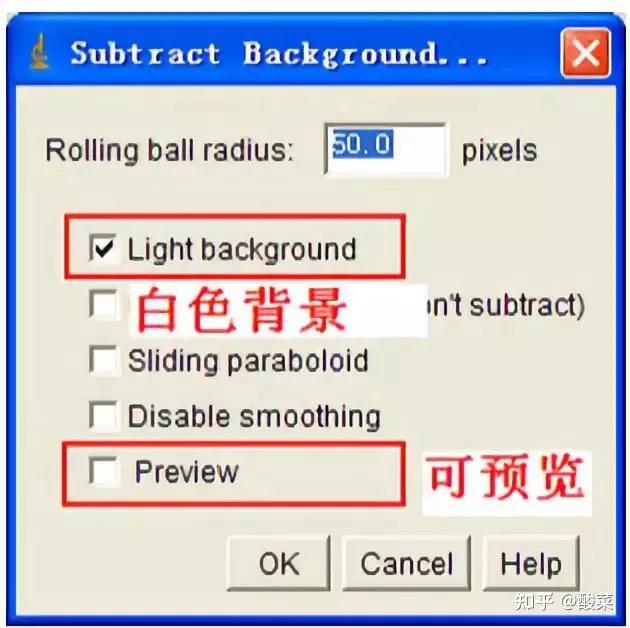
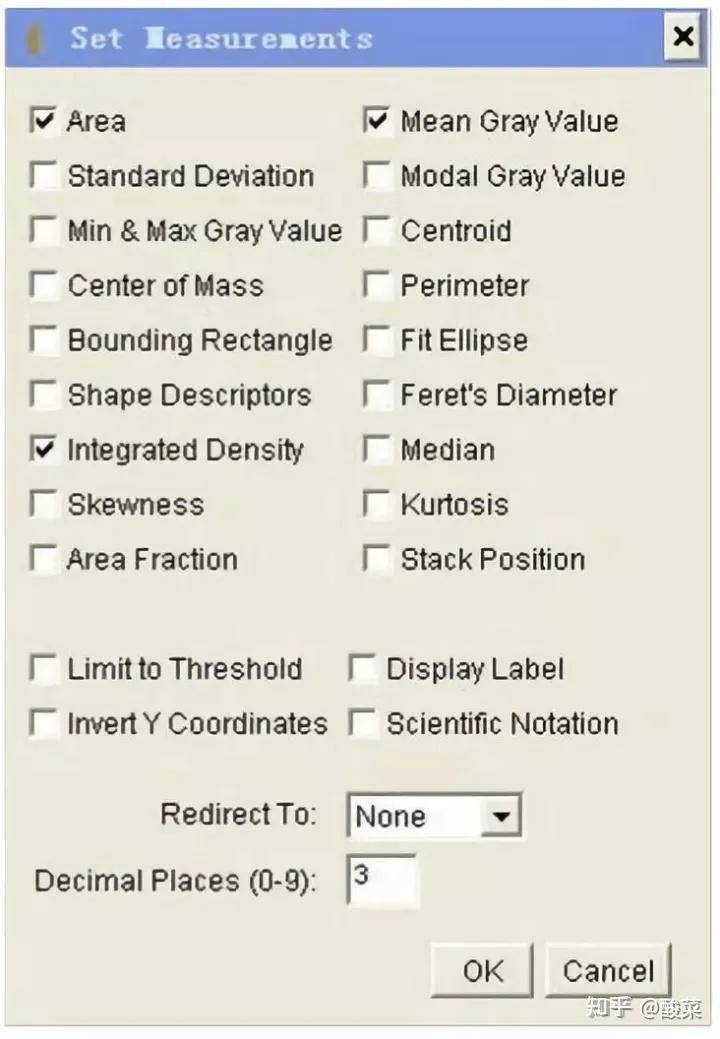
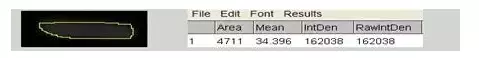
八、 凝胶图象分析
将胶片进行扫描或拍照,用image J等凝胶图象处理系统分析目标带的分子量和灰度值。
问题解决
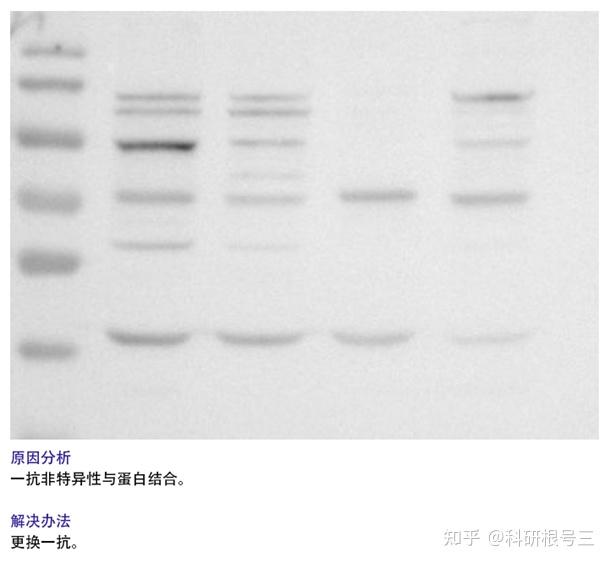
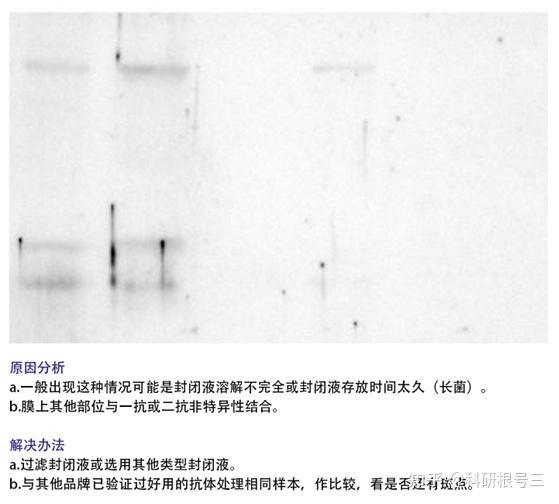
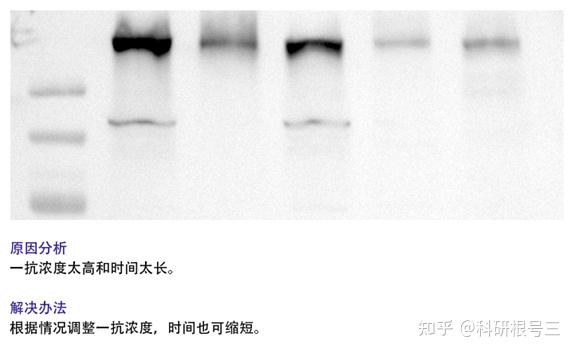
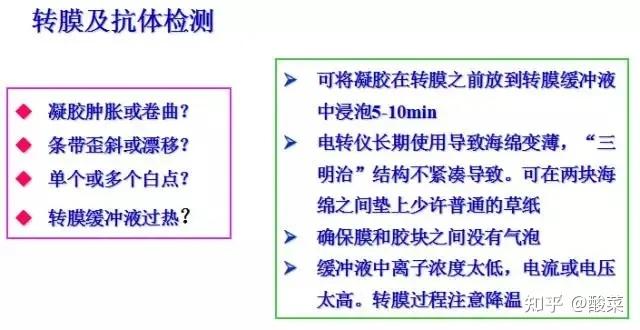
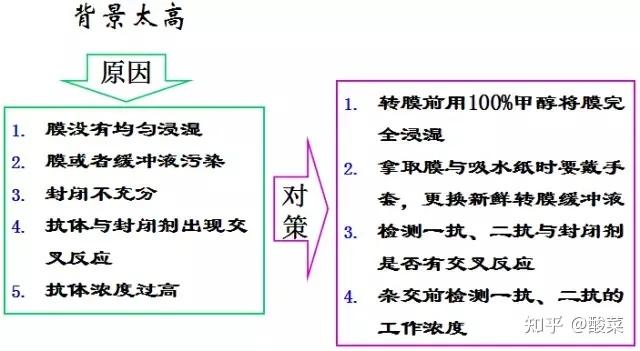
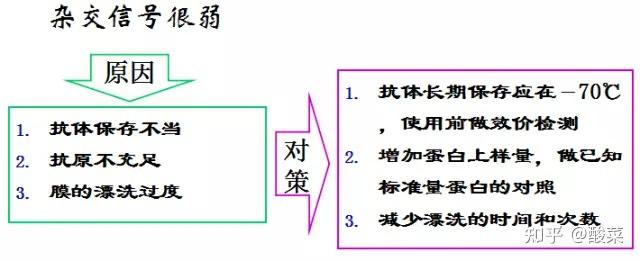
1. 检测到蛋白条带背景较深?
(1) 可适当增加TBST中的吐温浓度;
(2) 缩短二抗孵育时间;
(3) 增加牛奶封闭时间;
(4) 增加洗膜次数与时间;
(5) 降低抗体浓度;
(6) 转膜时注意用冰盒降温;
(7)更换抗体,选用特异性更好的WB抗体。
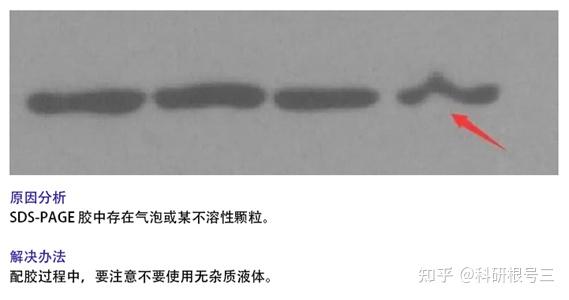
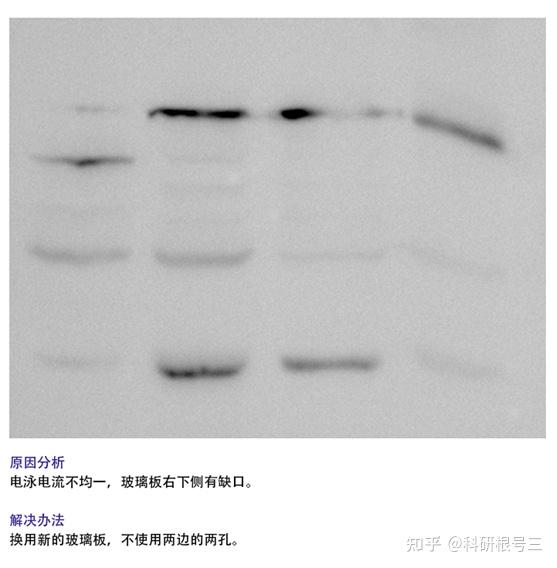
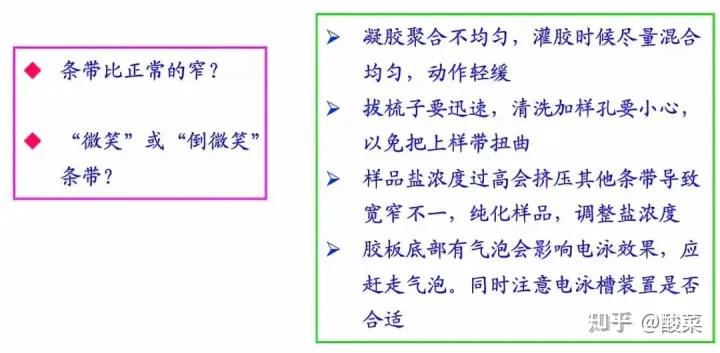
2. 检测到条带倾斜?
(1) 配PAGE胶时充分混匀;
(2)上胶时充分等到胶体凝固再电泳;
(3) 检查电泳盒中导电线是否水平;
(4) 降低上样总体积体系。
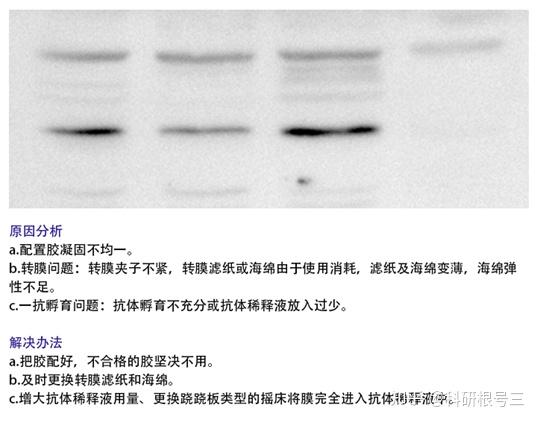
3. 目的蛋白分离不充分?
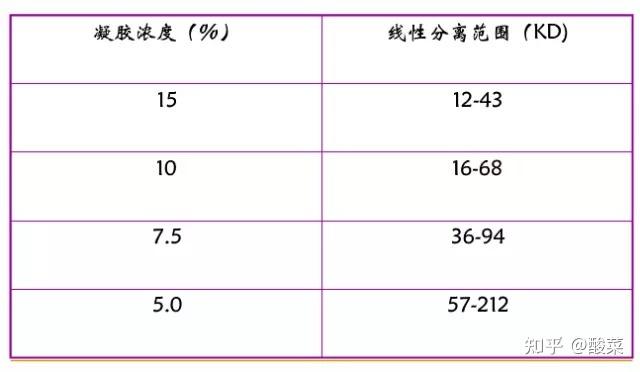
根据检测蛋白分子量,增加分离胶浓度与电泳时间。
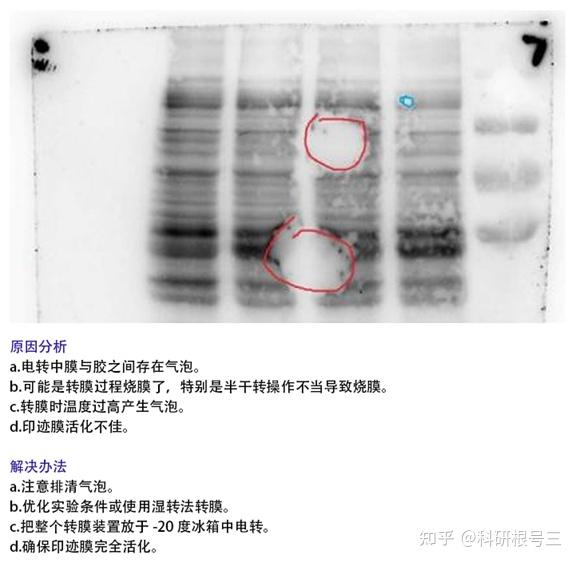
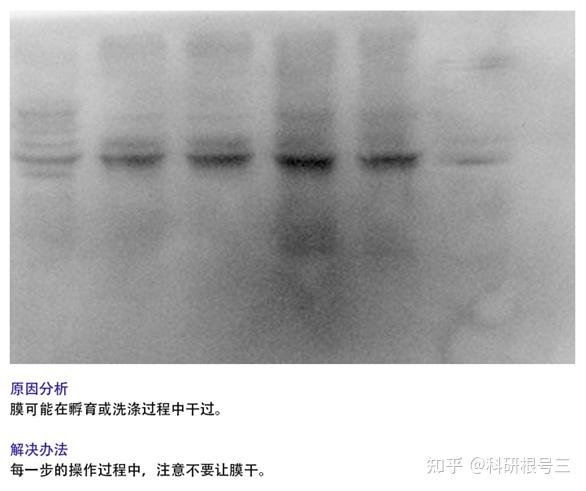
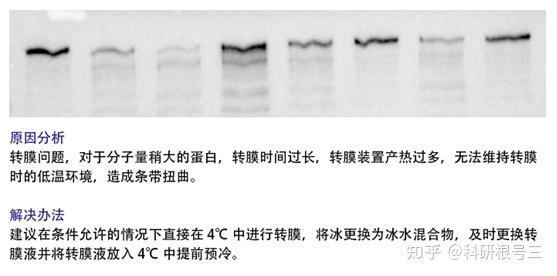
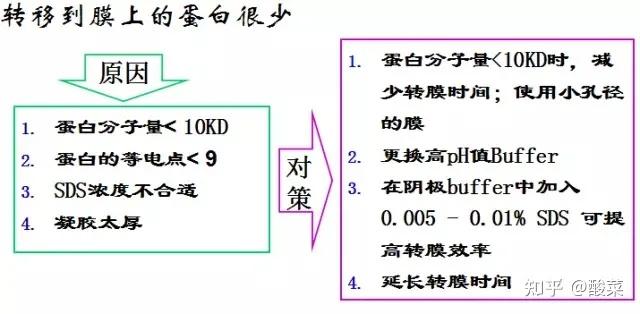
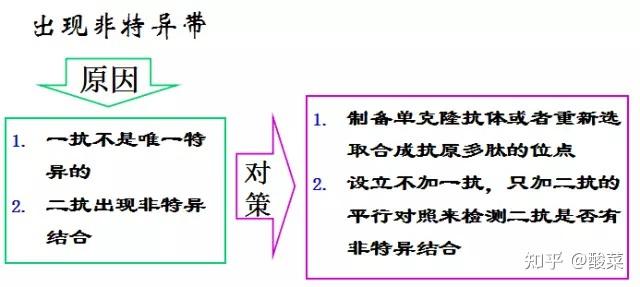
4. 未检测到蛋白条带?
(1) 蛋白样品制备后需要注意低温保存,防止蛋白降解;
(2) 确定上样浓度,调整上样体积;
(3) 转膜时确定电极是否正确;
(4) 转膜液重复利用次数太多,甲醇挥发较多;
(5) 确定AB荧光液充分混匀;
(6) 显色液应保证时间较新,显影时增加曝光时间。
参考文献:
1.WA, W., S, B., DA, O., HA, H., GM, F., CT, F., SM, C., and RA, Y. (2018). Author Correction: Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 562, E24.
2.Y, S., SD, J., Q, Z., L, H., J, F., XY, L., W, W., F, W., and RH, G. (2017). Long non-coding RNA LUCAT1 is associated with poor prognosis in human non-small lung cancer and regulates cell proliferation via epigenetically repressing p21 and p57 expression. Oncotarget 8, 28297-28311.
3.SK, K., H, L., K, H., SC, K., Y, C., SW, P., G, B., Y, L., JK, C., TK, K., et al. (2014). SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell stem cell 15, 735-749.
4.B, W., D, L., A, K., D, L., and O, K. (2014). Ionizing radiation-inducible miR-27b suppresses leukemia proliferation via targeting cyclin A2. International journal of radiation oncology, biology, physics 90, 53-62.
5.SS, M., LJ, V., N, R., JA, S., BM, F., J, M., KT, B.-R., J, L., D, I., MM, K., et al. (2017). A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer cell 32, 460-473.e466. |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-9-12 08:00
发表于 2024-9-12 08:00


 提升卡
提升卡