用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
侧向层析技术
›
TLC薄层层析技术
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
10334
|
回复:
0
TLC薄层层析技术
[复制链接]
非诚勿扰孟非
非诚勿扰孟非
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2024-9-4 07:36
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
薄层色谱,或称薄层层析(thin-layer chromatography),是以涂布于支持板上的支持物作为固定相,以合适的溶剂为流动相,对混合样品进行定性与定量分析、分离和鉴定的一种层析分离技术。
20世纪50年代从经典柱色谱法及纸色谱法的基础上发展起来的一种平面色谱技术;至20世纪60年代后,人们对薄层色谱法在使用器材的规格、操作方法及术语使用的标准化等方面进行了大量的工作,使该方法日趋成熟和完善,广泛地应用于有机合成中。
TLC薄层层析技术作为有机合成中,最直接的监测反应的手段,本文对其内容进行简单的介绍。
原理:
色谱法的基本原理是利用混合物中各组分在某一物质中的吸附或溶解性能的不同,或和其它亲和作用性能的差异,使混合物的溶液流经该种物质,进行反复的吸附或分配等作用,从而将各组份分开。
特点:
样品分离量少(微克~毫克级),分析快速。
分类:
按所用固定相材料不同,分为吸附、分配、离子交换、凝胶过滤等薄层层析。有机合成中常用的
硅胶,氧化铝薄层
属于吸附色谱。
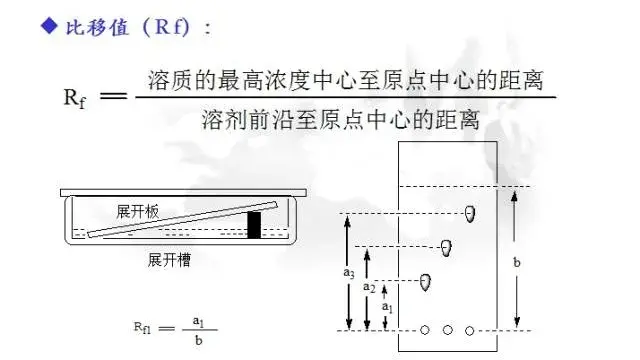
比移值(Rf):
化合物移动的距离大小用Rf值来表达。这是一个位于0~1之间的数值,它的定义为:化合物最高浓度中心距离原点基线(最先点样时已经确定)的距离除以溶剂的前沿距离原点基线的距离。
点样
----成功分离的关键
1、点样原点小而圆,直径尽量不要超过2mm(有机溶剂尽量用0.3mm直径毛细管,水等高粘度的溶剂用0.5mm直径的);
2、在便于显色的前提下,点样量尽量少,防止过载拖尾或掩盖Rf相近的点;
3、点样勿伤及薄层表面;
4、点样原点尽量远离薄层边缘,至少相隔3mm, 减少边缘效应(边缘经常跑歪);
5、选择规则的矩形薄板;
6、所有点样点要保持在一条与底边平行的直线上,标准点对照时,务必点交叉点;
7、点样完后,溶剂用电吹风尽量吹干。
展开剂的选择:
化合物在薄板上移动距离的多少取决于所选取的溶剂不同。在戊烷和己烷等非极性溶剂中,大多数极性物质不会移动,但是非极性化合物会在薄板上移动一定距离。相反,极性溶剂通常会将非极性的化合物推到溶剂的前段而将极性化合物推离基线。一个好的溶剂体系应该使混合物中所有的化合物都离开基线,但并不使所有化合物都到达溶剂前端,Rf值最好在0.2~0.8之间。虽然这个条件不一定都能满足,但这应该作为薄层色谱分析的目标(在柱色谱中,合适的溶剂应该满足Rf在0.2~0.3之间)。
常用展开体系极性大小顺序
石油醚<正己烷<二氯甲烷<四氢呋喃<乙酸乙酯<丙酮<正丙醇<甲醇<水
常用的混合体系
EA/PE(hexanes),DCM/MeOH
PE/DCM,PE/Acetone,Ether/DCM,EA/DCM,Acetone/DCM
展开剂的选择原则
1、对所需成分有良好的溶解性;
2、可使各成分间有较好的分离;
3、待测组分的Rf在0.2~0.8之间;
4、不与待测组分发生化学反应;
5、沸点适中,黏度较小;
6、展开后组分斑点圆且集中;
7、混合溶剂最好新鲜配制。
展开:
1、尽量用新配制展开剂(长时间挥发后比例会改变);
2、展开缸盖子密封性要好;
3、展开剂用量适中,不能漫过点样原点;
4、薄层板侧边不能贴到展开缸壁;
5、薄层板尽量用镊子夹住小心放入展开缸中,而不是用手捏住放入;
6、让溶剂向上展开约90%的薄板长度;
7、理想的展开位置为保证Rf值在0.2~0.8之内。
看板
----一看、二照、三碘、四显
1、从展开池中取出薄板并且马上用铅笔标注出溶剂到达的前沿位置, 根据这个计算Rf的数值;
2、用镊子小心取出薄层板,吹千溶剂;
3、首先直接观察,画出有色物质的斑点位置;
4、在紫外灯下观察有无暗斑或荧光斑点(并用铅笔画出斑点位置);
5、大部分有机物会吸附碘,可逆的产生棕色或黄色斑点;
6、千万注意,等高的点,不一定是同一个物质,有可能是极性相同的点,需要用其他监测方法(如LCMS)确定。
7、用破坏性方式观测薄板。当化合物没有紫外活性的时候,只能采用这种方法。使用染色剂时,将干燥的薄板用镊子夹起并放入染色剂中,确保从基线到溶剂前沿都被浸没。用纸巾擦干薄板的背面。将薄板放在加热板上观察斑点的变化。在斑点变得可见而且背景颜色未能遮盖住斑点之前,将薄板从加热板上取下。
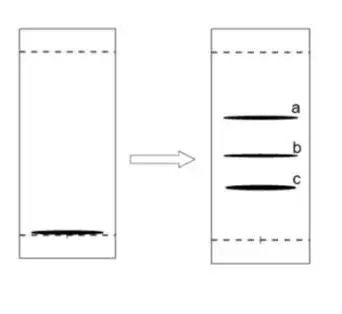
TLC-MS联用
用小板点成带状,分离少量的样品,刮取a、b、c三个纯点,MS检测出哪个点可能是我们的产物,取得标样,再展开prep-TLC得到纯化后样品,作 NMR鉴定。
TLC监测反应时机:
对于不稳定的化合物各个时机点最好都要监测。
1、反应半小时;
2、反应过夜前后,TLC检测并留样:
3、后处理前后,TLC检测并留样;
4、样品拌硅胶前后,TLC检测并留样;
5、机分样品浓缩冻千前后,TLC检测并留样。
TLC为柱分离寻找条件
1、选择不同的混合体系, 多次尝试,如果想让Rf变得更大一些,可使溶剂体系极性更强些;如果想让Rf变小,就应该使溶剂体系的极性减小些。如果在薄板上点样变成了条纹状而不是一个圆圈状,那么你的样品浓度可能太高了。稀释样品后再进行一次薄板层析,如果还是不能奏效,就应该考虑换一种溶剂体系。
2、找到目标物Rf值0.2左右,并与其他杂质分离度较大的系统;
3、然后根据样品分离度,硅胶规格,柱径比,硅胶与样品比例等综合信
息,选择比Rf值0.2左右体系稍小极性的体系进行柱层析。
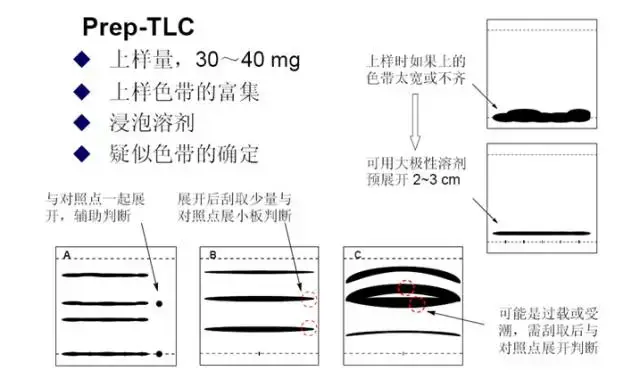
Prep-TLC分离样品
:
样品要求:因为硅胶板是弱酸性的,对酸不稳定的化合物尽量避免爬大板;对空气,水,有机溶剂稳定; 低沸点的溶剂里要好溶;最好有紫外吸收;极性不能太大(至少展开剂能爬起来)。
上样:尽量少的溶剂溶解样品,最好是易挥发的,二氯甲烷最常用,如果不溶可以加几滴甲醇促溶。可以用一次性滴管塞棉花后剪成毛笔状上样,每个人的办法都不同,小编就是这么弄的,顺便练练毛笔字。上样时也最好像写毛笔字一样一气呵成,这样跑出来的带比较均匀。如样品溶液太多需要上两遍样,先用吹风机吹干溶剂后再上样。
跑板:展开剂的选择,先用小板爬样,选到合适的展开剂后,配好溶剂爬大板,由于大板和小板由细微的差别,大板展开剂的极性可以稍微比小板展开剂的极性大一点,一样的话也问题不大。跑板最好要跑到快顶端1cm左右,充分展开样品,切记不要跑过,跑过的话,极性小的点可能会被展开剂冲都一起,而极性大的点虽然不会冲到上面,但会变散,不容易判断色带的边缘。
检测:产品的判断,通过极性大致判断哪几个色带可能是产品,然后每个色带可以先刮一点,碾碎,加甲醇溶解,过滤送LCMS检测判断。
刮板:判断好色带后,在紫外灯下,用铅笔描好荧光带,开始刮板。带好口罩很重要,小编是用美工刀刮的,刮不干净的可以用小板刮可以刮干净。
洗脱:挂好的硅胶,量少的话,可以隔着称量纸用试管擀碎。量多的话,直接装到自封袋里面随便蹂躏,直到成粉末为止。粉末状的硅胶样品的洗脱,大部分人是直接加溶剂泡出来。
但小编用的是,把硅胶粉直接装到柱子(有底层砂芯的那种)里,上层铺一薄薄的石英砂,直接像过柱子一样在上层加溶剂压出来,这种方法的好处就是,对于溶解性不好的样品可以快速洗出,用相对较少的溶剂洗出产品。加入溶剂洗脱的时候可以随时点板看产品是否已经完全洗出。
常见拖尾原因及解决办法
1、首先考虑的是样品浓度过大,薄层板过载导致。这种情况直接降低样品浓度或者是上样量就可以验证了。
2、样品对硅胶的吸附能力过强导致的拖尾。对不同体系加入不同的调节剂,酸体系加冰醋酸或甲酸,碱体系加氨水、三乙胺或二乙胺。
3、展开剂的极性与样品极性不附,不能做到有效展开导致。可以通过调节展开剂极性解决。
4、如果是长带状,那最可能的原因是展开剂对样品的溶解度不够所导致。可以根据极性表换极性相近的对样品溶解度更好的溶剂做展开剂,另外样品未溶解完全,点在班上的样品有未溶固体样品也会导致长条状拖尾。
原文地址:https://zhuanlan.zhihu.com/p/468171497
回复
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
浏览过的版块
百科词条
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-4 07:36
发表于 2024-9-4 07:36