登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
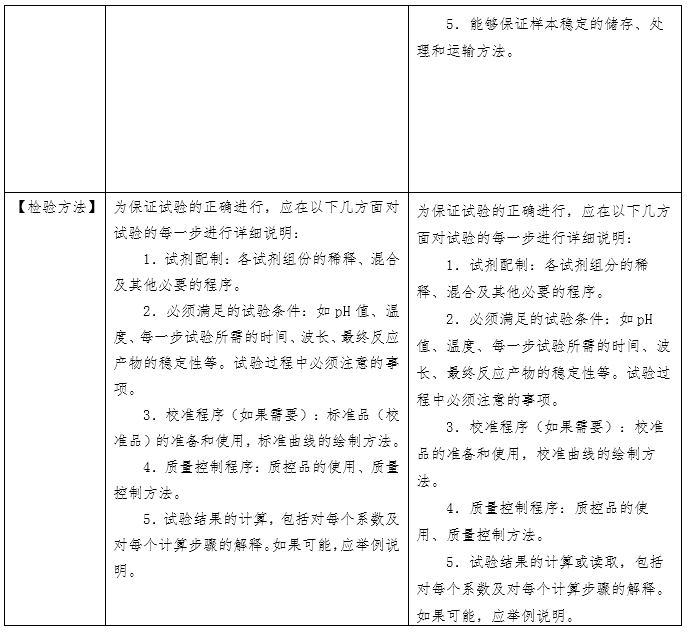
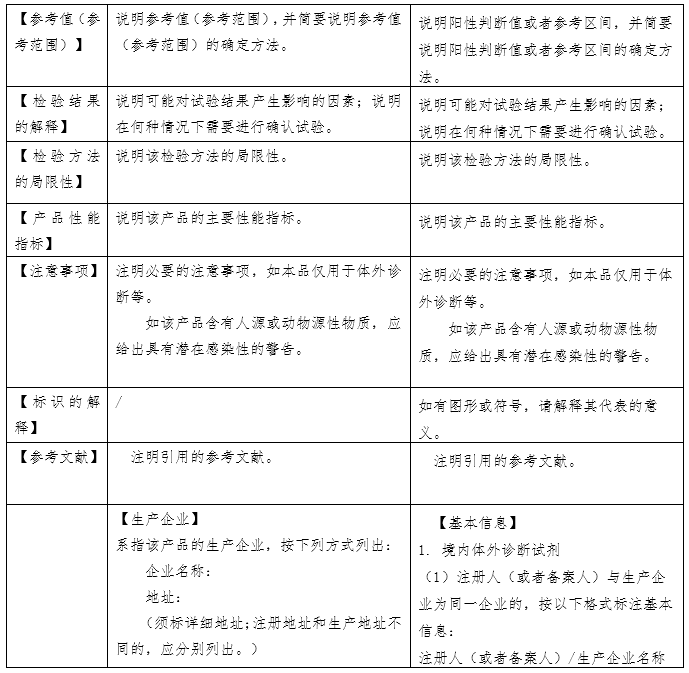
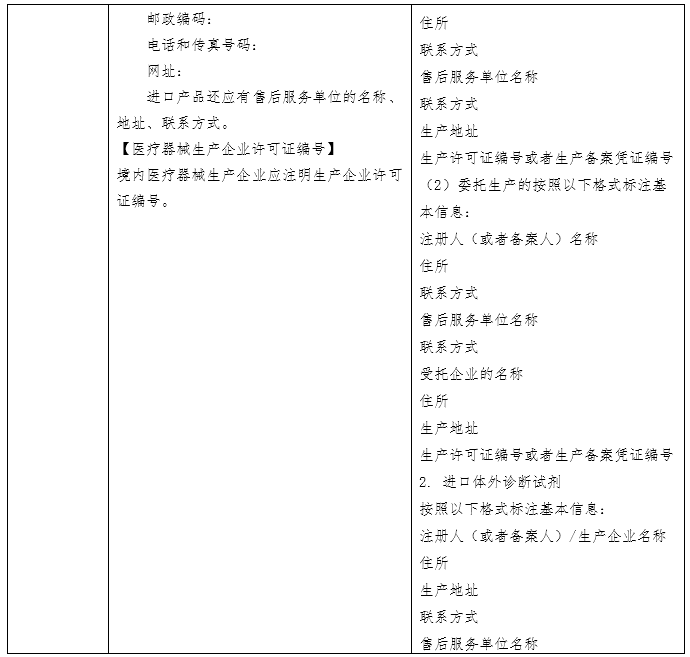
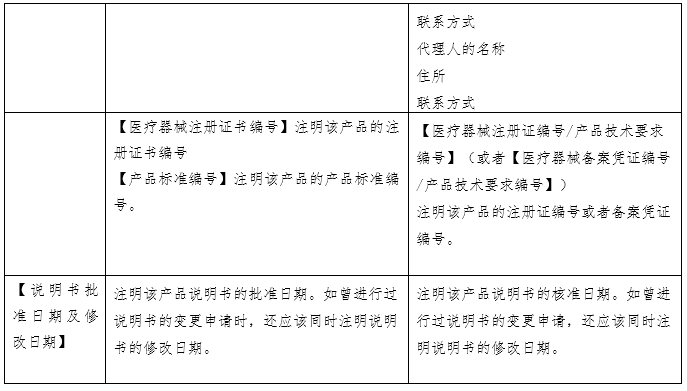
(原创 2023-06-16 CMDE 中国器审) 当我们选购日常食品时,会看产品的名称、产地、配料表、生产日期、储存方式,从而判断食品是否含有过多的食品添加剂或者脂肪含量是否过高、哪个产地的生产环境和监管环境更好、生产日期是否新鲜,买回家后如何保存。在整个环节中,食品标签展现了两个功能:一是展示产品从而被目标客户选中;二是指导客户使用和保存。 体外诊断试剂作为商品,其说明书也具有上述“食品标签”的功能,一是展示产品的性能,二是指导客户使用和保存。但相比日常食品,体外诊断试剂的检测结果直接关系到疾病性质的定位、病情程度的把握、治疗方案的选择及治疗预后的评判,与民众生命安全和健康关系重大,其安全有效性显得尤为重要。因此,作为一种特殊的商品,体外诊断试剂受到全生命周期的严格监管。而说明书作为注册批件的一部分,是监管过程中的重要依据和工具。综上,体外诊断试剂说明书(以下简称说明书)具有三个功能,一是展示产品的性能、二是指导客户使用和保存、三是监管的依据和工具。 国家药品监督管理局于2007 年先后发布了《体外诊断试剂注册管理办法(试行)》、《体外诊断试剂说明书编写指导原则》等系列文件。2014年发布了《体外诊断试剂注册管理办法(国家食品药品监督管理总局令第5号)》、《体外诊断试剂说明书编写指导原则(2014年第17号)》。2007版说明书指导原则参考了国际临床化学联合会关于体外诊断试剂盒标签和说明书的书写要求,2014版说明书则在2007版的基础上修改(下表为两版说明书的对比)。两版说明书编写指导原则规范了各种类体外诊断试剂产品说明书编写的框架和基本内容,具有普适性。同时,具体产品的指导原则如《新型冠状病毒(2019-nCOV)抗原检测试剂注册审查指导原则》在《体外诊断试剂说明书编写指导原则》的框架下,结合产品的具体特点,对此种类产品的说明书编写做出了特殊性和针对性的要求。
近年来体外诊断试剂产业飞速发展,产品种类不断丰富,检验方法逐步从纯手工法到半自动化、全自动化,自测产品需求不断增加,产品更新换代加快等,都为说明书的指导原则编写带来挑战。
一、全自动检测系统的普及使得【检验方法】项下需编写的内容发生改变。在手工法或是半自动化时期,需检验人员依据说明书【检验方法】中列出的操作步骤进行试剂的配制、混合、在固定温度孵育、冲洗等,接着人工输入检测仪器参数的设置、绘制校准曲线,得到检验结果或者需要依据仪器显示的数字进行人工计算。而全自动检测系统只需在设置好反应参数后将样本放到固定位置,检验人员无需手动操作便可获得检验结果。在这种情况下,全自动检测系统中仪器的参数设置反而是检验人员需要和关注的。此外,从监管的角度出发,体外诊断试剂的反应体系需要进行重点关注。因此,【检验方法】项下内容应体现样本的用量、试剂各组分的用量、反应参数的设置。 二、【主要组成成分】项下需载明的主要组成成分有待明确。目前,监管方、申请人和使用者基于关注点不同,对哪些成分属于主要组成成分的理解不同。同时申请人基于对申报产品配方的保护,会隐去部分组分,如抗生物素抗体、抗体阻断剂、缓冲液的特殊配方等。这就造成了审评过程中大量针对此项内容书写规范的发补。因此,需要基于目前的产业情况,平衡知识产权的保护和监管需求,明确说明书【主要组成成分】项下需载明的具体内容。 三、校准品和检测试剂的不匹配问题。检测试剂本身并不具有量值的赋值功能,需依赖校准品所绘制的校准曲线对检测样本进行赋值。生产厂家在试剂研发过程中,依照相应的指导文件建立量值传递的溯源链,逐步将有证参考物质、参考方法或企业参考品的量值传递至产品校准品,并计算校准品的不确定度。整个过程属于一个封闭系统,试剂和校准品固定搭配使用。但在实际应用中,部分厂家会提供无证的校准品和质控品配合检测试剂使用,或者A厂家的试剂配套未验证过的B厂家的校准品使用。《体外诊断试剂注册与备案管理办法》规定,校准品和质控品可随检测试剂盒申报,也可单独申报。结合近些年的审评经验,越来越多的企业基于商业便捷性和可操作性考虑,会选择单独注册申报校准品和质控品。当试剂和校准品/质控品分开进行注册申报时,若无法进行有效的关联,便会存在脱钩现象。因此,需在检测试剂、校准品和质控品各自的说明书中明确配套使用的其他试剂及其注册证号(备案号)和货号,固定检测系统,为使用人员提供参考。 四、【适用仪器】项的书写规范仍需提升。检测试剂的性能需联合检测仪器一同体现。我国体外诊断用仪器按照医疗器械管理,与检测试剂分开进行注册申报。一张仪器注册证中可包含多个型号,各个型号之间的差异有可能仅为外壳颜色的不同,也有可能是反应模块的删减和改进。所以,目前要求适用仪器的书写应明确仪器型号。但对于同一型号,均可由一个集团下的多个公司如郑州安图和北京安图,或者德国罗氏和苏州罗氏生产。此时,便会面临适用仪器的注册人不同的问题。目前在实际执行中,申请人大多以“安图”“罗氏”等类似的简称进行书写。而此项内容是否需要强调适用仪器的注册人仍需慎重考虑。 五、【产品性能指标】项下内容的完整度不够。随着《定量检测体外诊断试剂分析性能评估注册审查指导原则》、《定性检测体外诊断试剂分析性能评估注册审查指导原则》、《体外诊断试剂参考区间确定注册审查指导原则》、《免于临床试验的体外诊断试剂临床评价技术指导原则》、《体外诊断试剂临床试验技术指导原则》等通用性指导原则的发布实施,体外诊断试剂的评价指标逐渐完善。但在实际审评过程中,申请人并未意识到说明书【产品性能指标】项下内容是分析性能评估和临床试验的总结内容,需体现研究过程和结果。因此目前大多数申报产品在首次提交时编写过于简单。对此,说明书编写指导原则应针对【产品性能指标】项下的书写内容予以丰富和明确。 六、说明书的可读性和针对性有待提升。目前我国批准的体外诊断试剂可大致分为专业人员使用的专业产品和普通民众使用的自测产品。两类产品的使用者不同,关注点不同、对书写内容的理解能力也不同,因此说明书的编写重点也应有侧重区分。但目前专业产品和自测产品共用一套说明书编写指导原则,导至了说明书的可读性下降。另外,校准品和质控品有其独特性,且与检测试剂相比书写项目较少,适宜与检测试剂的说明书编写有所区分。 七、基因测序和人工智能判读软件所带来的监管挑战。基因测序产品可产生几十G的数据量,测序结果可编成一本薄书,所依赖的数据库需不定期更新。人工智能判读软件会在使用过程中不断完善算法,更新软件版本。面对这两种基于大数据的产品,说明书如何发挥应有的指导用户使用、展示产品性能和成为监管工具的功能仍需探索解决。 基于上述问题,《体外诊断试剂说明书编写指导原则》于2022年启动修订,对现行2014版指导原则公开征求意见,形成了2022年修订版初稿,经公开征求意见、企业座谈会后修改形成送审稿。需要说明的是,基于现行法规和当前认知水平,并考虑过渡稳定性,2022年修订版针对上述部分问题提出了解决方案,仍有部分问题需要随着产品应用发展不断进行解决和完善。
参考文献:
[1]吴琨.浅析体外诊断试剂产品说明书的常见问题[J].中国医疗器械信息,2011,17(11):18-22.DOI:10.15971/j.cnki.cmdi.2011.11.007. [2] 张丽.国际临床化学联合会关于体外诊断试剂盒标签和说明书的书写要求[J].中国新药杂志,2001(10):785-786. [3] 张丽. 有关体外诊断试剂说明书、包装、标签撰写的建议[C]//中华医学会微生物学与免疫学分会.第6次全国微生物学与免疫学大会论文摘要汇编.第6次全国微生物学与免疫学大会论文摘要汇编,2004:44-47. [4] 张丽.再谈体外诊断试剂标准及说明书的起草撰写[J].中国新药杂志,2011,20(19):1846-1847. [5] 李晓,张欣涛,郝擎,朱炯,洪伟.2020年国家医疗器械抽检体外诊断试剂品种质量状况分析[J].中国医疗器械杂志,2022,46(04):459-463. [6] 杨笑鹤,董沁芳,朱文武,甄辉.第二类临床化学体外诊断试剂(盒)注册申报常见问题分析[J].中国医疗器械杂志,2020,44(06):537-540+557. [7] 杨笑鹤,马琳榕,王雯,甄辉.体外诊断试剂产品全生命周期监管常见问题分析与建议[J].中国医疗器械杂志,2018,42(02):129-132. [8] 马忠明,杨波.我国体外诊断试剂监管现状、面临的新形势及其思考[J].中国食品药品监管,2022(06):4-9. [9] 梁爽,孙轶康,仲志真.上海市第二类体外诊断试剂注册申报常见问题和对策分析[J].中国医疗器械信息,2023,29(03):10-12+45.DOI:10.15971/j.cnki.cmdi.2023.03.040. [10]张龚敏,赖锦坝,李风梅,何泽文.医疗器械产品说明书编写规范探讨[J].中国医疗器械信息,2019,25(03):3-4+78.DOI:10.15971/j.cnki.cmdi.2019.03.002. [11]李哲,姜海鑫,王京科,沈洁芳.体外诊断试剂在使用环节的法规风险探讨[J].中国医疗器械杂志,2018,42(01):62-63. [12]章宏法,张敏红.医疗机构体外诊断试剂使用管理现状调查及思考[J].中国医疗设备,2010,25(11):10-13. [13]范仁祥.体外诊断试剂现状剖析[J].中国药事,2006(11):663+690. [14]范仁祥.体外诊断试剂监管中存在的问题及对策探讨[J].中国药业,2006(11):10-11. [15]张华荣.体外诊断试剂的正确选择和使用[J].检验医学与临床,2004(02):84-85. 雷山 审评六部 供稿
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2023-11-13 15:04
发表于 2023-11-13 15:04