|
《医疗器械注册管理办法》(总局令第4号)及《体外诊断试剂注册管理办法》(总局令第5号)实施后,相关行政许可事项的审评审批时限有增有减。这一点我们在之前的推送中为您进行了详细的对比。 依据2014版《注册管理办法》,一般的审评审批流程包括:受理、资料承转、技术审评、再次技术审评(针对补充资料)行政审批、制证发布。今天我们为您详细梳理申请不同行政许可事项时,面临的不同流程,各环节时限与总时限。 特别需要说明的是,此次发布的内容包含了对之前发布内容的勘误,主要针对“延续注册的审评审批流程及时限”部分。由于《注册管理办法》未明确延续注册技术审评时限,因此依据受理中心与器械注册司对此疑问的口头答复制定下表。希望该内容对大家有所帮助! 上述的一般审评审批流程及相应时限未包括以下情况: 1.需要外聘专家审评的, 2.药械组合产品需与药品审评机构联合审评的, 3.审评期间组织对申请人进行与产品研制、生产有关的质量管理体系核查的。 另外,2014版《注册管理办法》对于“境内”及“境外”产品在“审评审批时限”方面的规定无区别。因此,下列表格对于境内及境外产品均适用。 以下是详细内容(注:表格中标记“—”表示该流程不包含本环节):
第III类医疗器械或体外诊断试剂产品 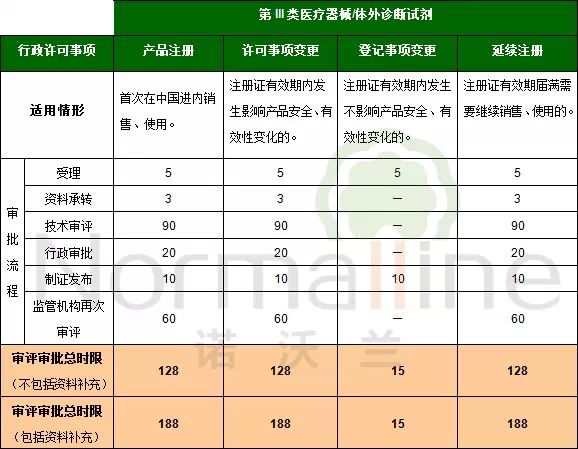
第II类医疗器械/体外诊断试剂 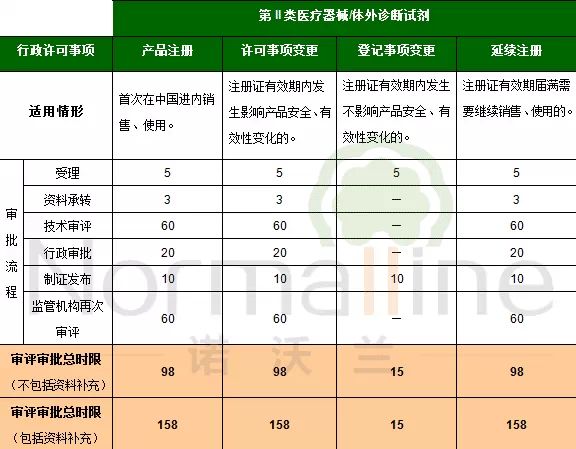
第I类医疗器械/体外诊断试剂 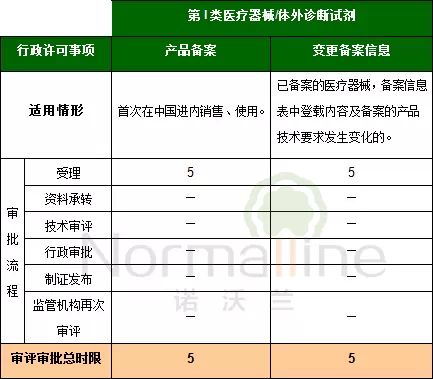
|  [复制链接]
[复制链接]

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2013-7-12 23:23
发表于 2013-7-12 23:23



 提升卡
提升卡



