金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
写在前面的话
2021年初读到的浙江大学药学院范晓辉教授团队发表的一篇关于“空间转录组技术”的综述。记得年初的时候,找空转的综述相对还是较少的,现在已经有非常多了,公众号的解读文章也越来越多。当时初读囫囵吞枣,今天计划再次仔细地复习一下。重点会放在辨别几种空间转录组测序策略的概念区别上。
文献基本信息
Title:Uncovering an Organ’s Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics
标题:利用空间分辨转录组学揭示单细胞分辨器官的分子结构
Journal:Trends in Biotechnology(Cell 系列)
IF:14.34
发表时间:2021.01
单位:浙江大学 药学院 药学信息研究所
第一作者:Jie Liao
通讯作者:Xiaohui Fan (范晓辉)
文章类型:Review
DOI: https://10.1016/j.tibtech.2020.05.006
Abstract:生物学和药理学研究的基础之一,是揭示组织空间状态下功能与结构精细的细胞异质性。与传统的单细胞或普通转录组不同,最新的空间转录组技术提供了接近单细胞或亚细胞分辨率下的组织表达信息。跨尺寸的大规模信息结合位点标签可以更好地展现组织3D转录组图谱。本综述深入讨论最常用的空间分辨转录组学分析方法、技术及其在基础和生化研究中的巨大应用潜力。

文献信息
文章主体导图
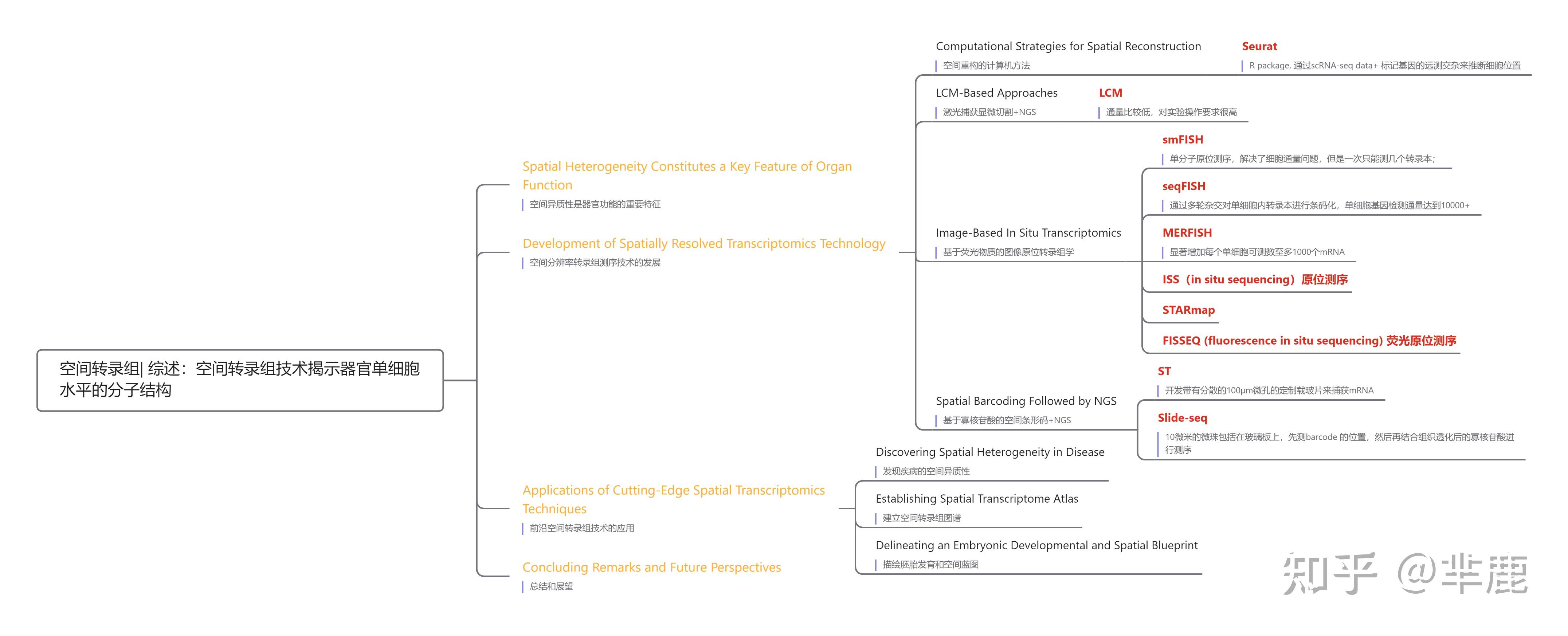
文章主体思维导图
文章重点解读
1 空间异质性是器官功能的重要特征
人体组织是一个高度复杂的系统,由多达数万亿个细胞组成,这些细胞在种类、时间和空间上各不相同,但相互协调,形成独特的微环境,以维持器官功能和处理信息。人体器官分子基础研究从初级到深层次都有涉及,通过这一广泛的过程,人们逐渐达成科学共识,即无数具有不同功能类型的细胞数量,加上发育(时间)和区域(空间)差异,构成哺乳动物器官转录异质性的主要组成部分。
高通量单细胞RNA测序(single-cell RNA sequencing,scRNA-seq)的新兴前沿技术在推断细胞类型和轨迹方面取得相当大的成功,特别是将scRNA-seq方法应用在发育生物学问题。伪时间分析、标记细胞起源的内源性条形码等方法是在一定时间间隔连续取样以揭示细胞命运、细胞谱系和发展轨迹,这些信息一旦在悬浮状态下解离,就需要进行基于流动细胞(原生质体)的scRNA-seq,如inDrop、Drop-seq、Microwell-seq和SPLiT-seq。
空间转录组学一直是研究人员的长期目标,从早期尝试使用基于荧光原位杂交(fluorescence in situ hybridization,FISH)的方法到目前单细胞分辨率的空间转录组检测。最近,有研究者利用他们较早提出的RNA成像方法——多重抗误差矫正荧光原位杂交技术(MERFISH)呈现下丘脑视前区的空间分辨图谱,根据其研究结果,下丘脑视前区大约有70簇神经元,远小于整个大脑。同一群体内的细胞在空间分子特征相互区别,为了更好地理解细胞,需要同时记录它们的转录异质性和空间坐标。
本综述深入讨论最常用的空间分辨转录组学分析方法、技术及其在基础和生化研究中的巨大应用潜力。
2 空间分辨转录组学技术的发展
按照组织分辨率分,空间转录组技术可以分为三类:
按照实验技术策略来分,空间转录组技术可以分为四类:
- 基于组学实验结合空间重建的计算策略:
- 激光切割与NGS测序结合的策略:
- 基于荧光物质原位的转录组学;
- 基于寡核苷酸的空间条码加上NGS测序;
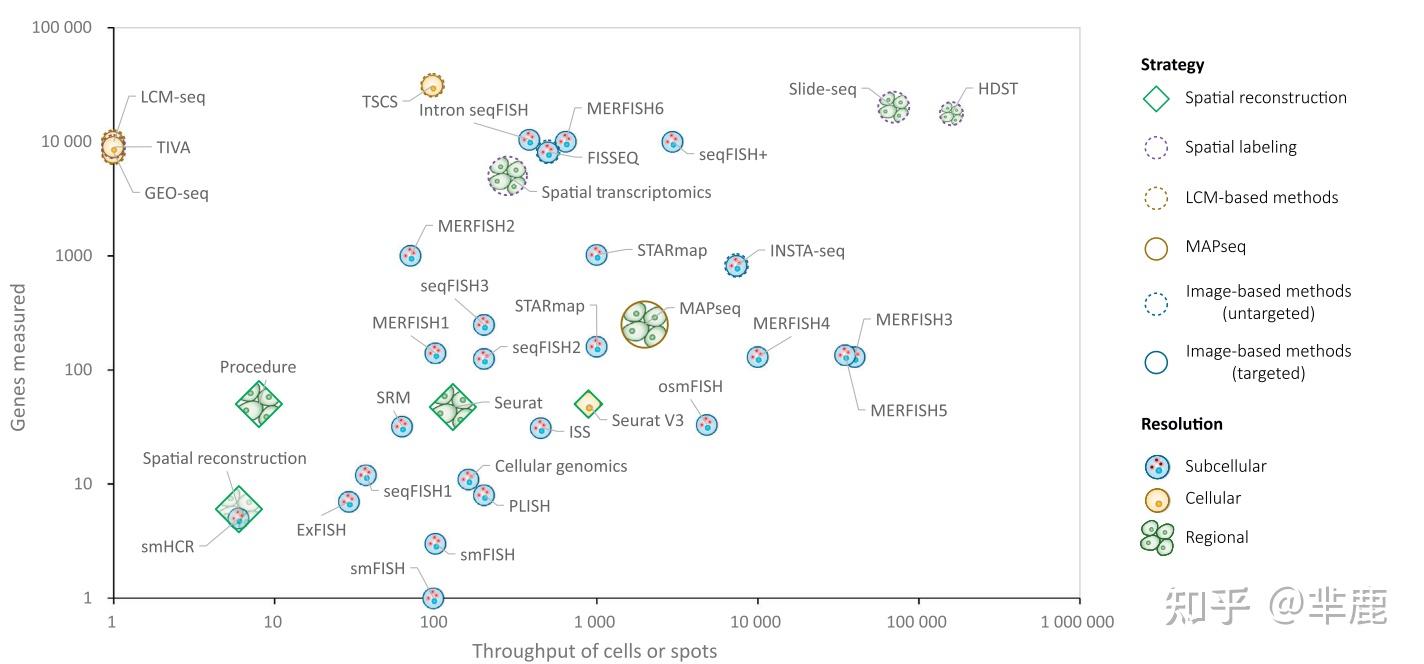
每种空间分辨转录组方法的基因和细胞通量
方法按照其类别,以不同的形状、符号和大小呈现
菱形:用于空间重构的电子方法,紫色圆:空间标注方法,橙色圆:基于LCM的方法,橙色圆:MAPseq,蓝色圆:基于图像的目标方法,蓝色圆:基于图像的非目标方法;
不同符号代表每种方法的空间分辨率(橙色细胞:细胞分辨率,蓝色细胞:亚细胞分辨率,多细胞:区域分辨率)。每个符号的大小大致与其空间分辨率有关。
x轴和y轴分别表示每柄被测细胞或点的数目和他们实验中确定的实际基因。
简称:ExFISH:扩展FISH、FISH:荧光原位杂交、FISSEQ:荧光原位测序、GEO-seq:空间位置测序、HDST:高清空间转录组学、ISS:原位测序、LCM:激光捕获显微切割、MERFISH:多重抗误差矫正荧光原位杂交技术FISH、MAPseq:基于测序技术的多重分析,osmFISH:ouroboros单分子FISH,PLISH:邻近连接原位杂交,secFISH,序列FISH,smHCR:单分子杂交链式反应,SRM:超分辨显微镜,STARmap:空间分辨转录放大读出图,TIVA:体内转录组分析、TSCS:空间单细胞测序。
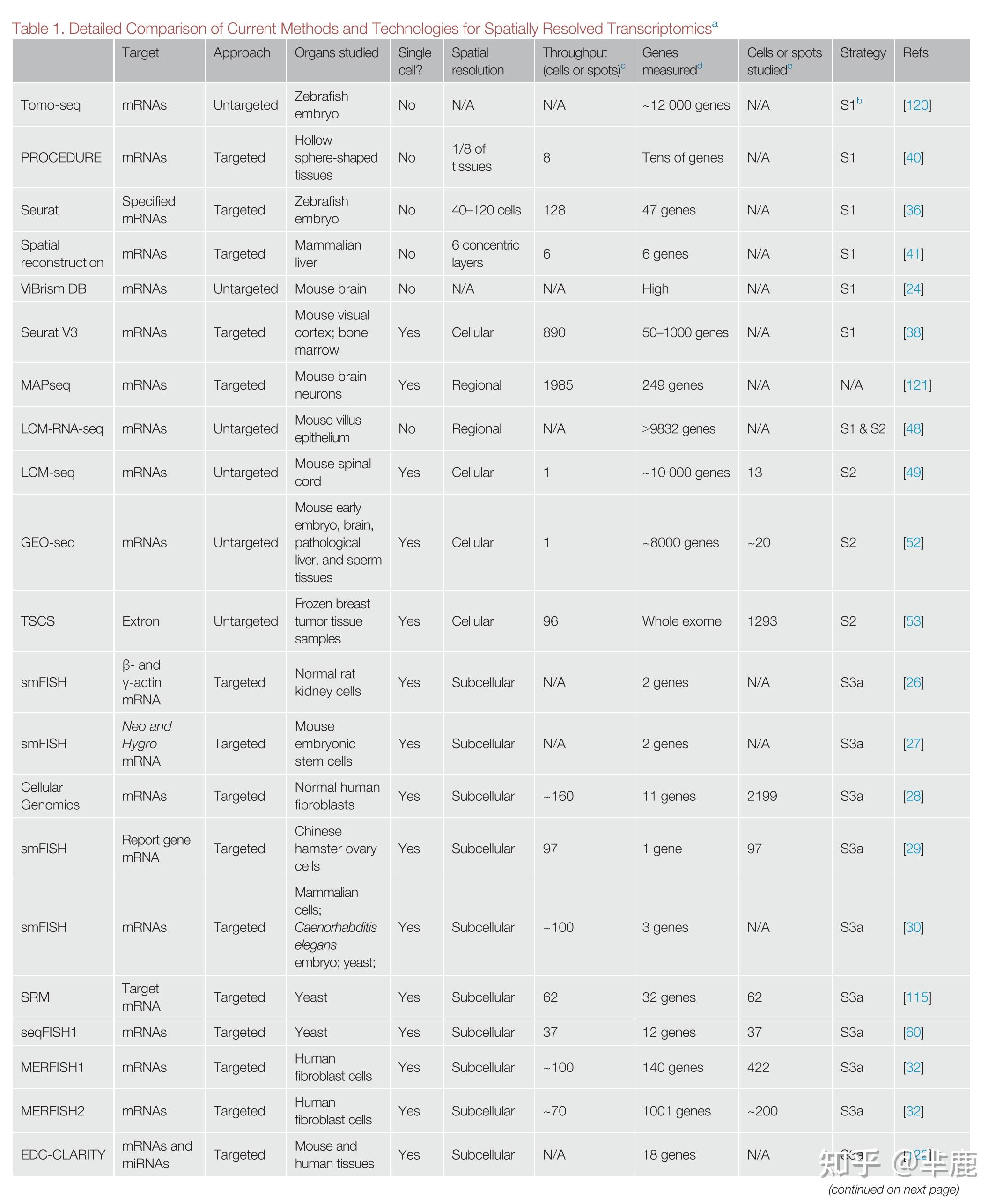
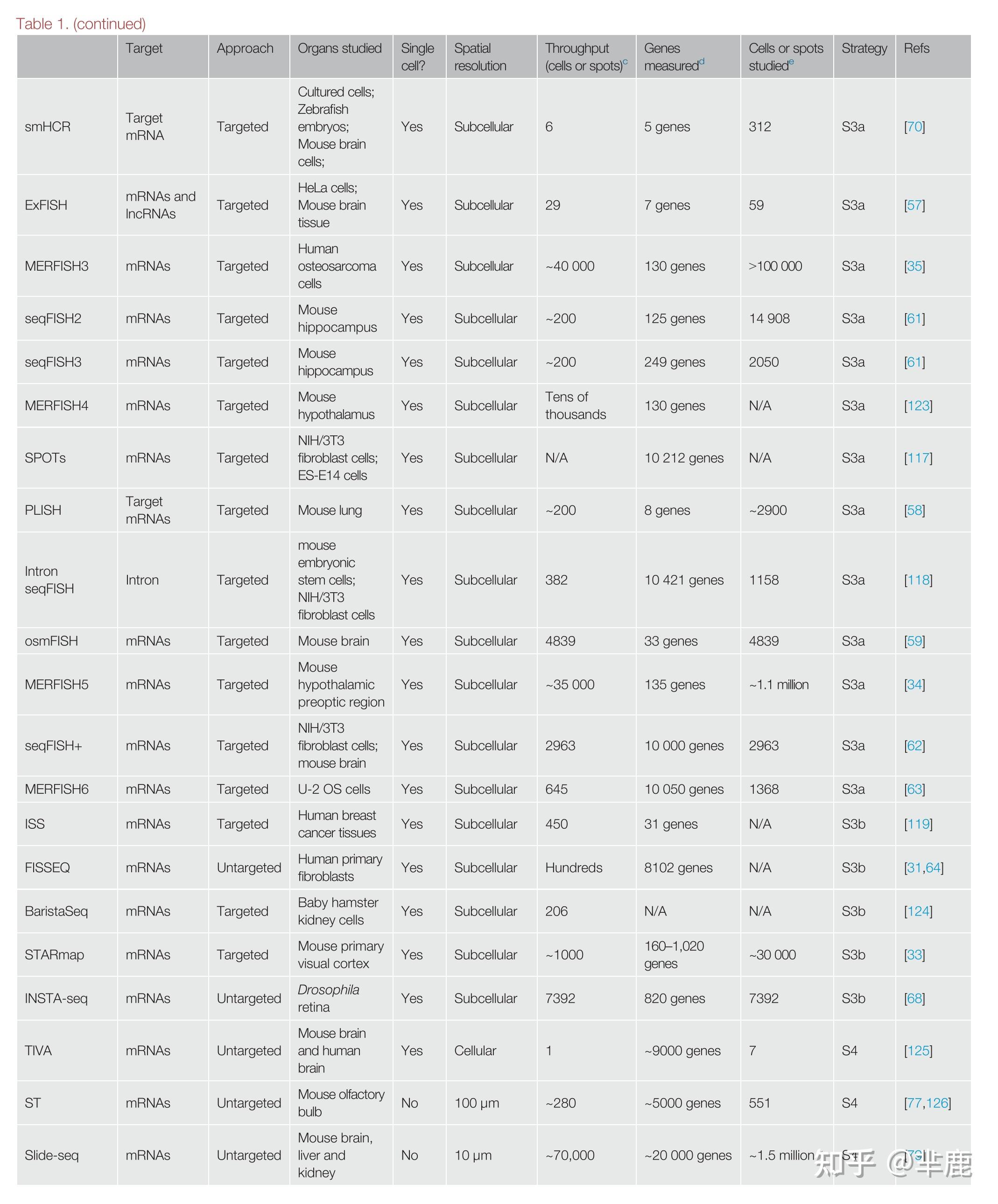
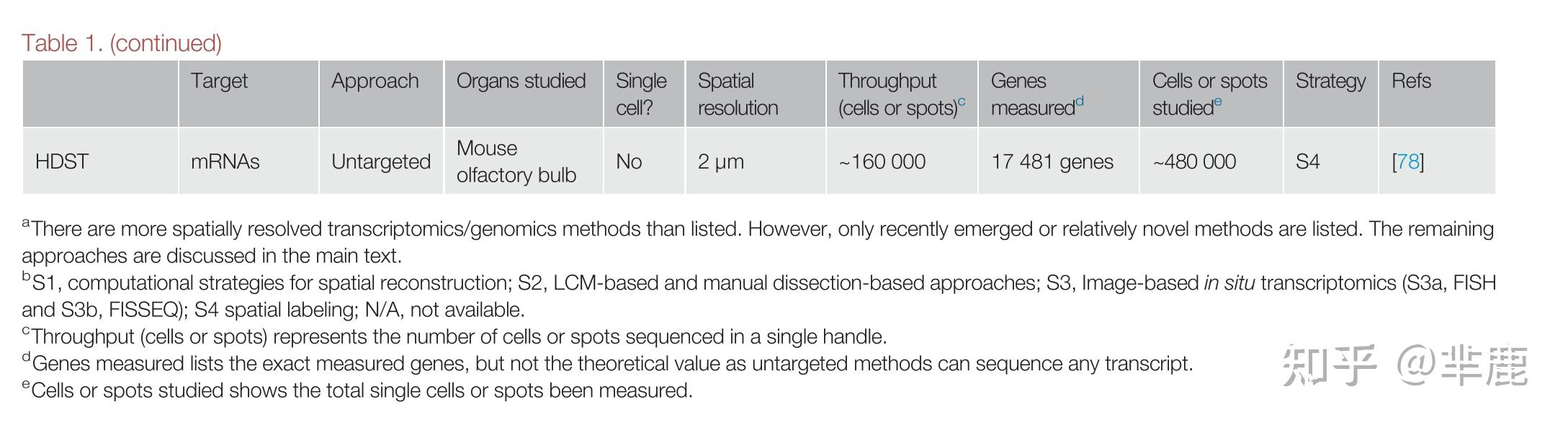
详细比较目前空间分辨转录组学的方法和技术
2.1 空间重构的计算机方法
由于流式细胞scRNA-seq过程中细胞空间信息丢失,RNA染色方法只检测到部分转录本,因此整合多尺度数据,将电子技术应用于组织的空间转录组重建是必要的。例如,Satija和同事开发Seurat,这是一个广泛分布的R包,可以通过结合scRNA-seq数据和标记基因的原位杂交来推断细胞定位。具体来说,SMART-seq对发育中的斑马鱼胚胎851个个体细胞的单细胞基因表达进行分析,将细胞分为不同类型,并为每个细胞类型产生标记基因。标记基因FISH实验显示它们表达模式的空间分布,产生标记基因在整个组织的表达密度。最后,根据标记基因的表达谱和表达密度可以定位单个细胞。
局限性:这种计算机方法充分利用固有的基因表达模式或共表达趋势,提供从单细胞的巨大转录组学数据中获取细胞与细胞相互作用的信息,但这些演绎法在某些情况下受到限制,只呈现空间趋势或特定组织的总体布局。因此,有必要精确描述组织中实际的单细胞转录组织,开展更深入的分析。
2.2 基于LCM的方法
另一种获取精确位置信息的转录组的方法是利用LCM从组织切片中单独分离单细胞或整个感兴趣区域。LCM是一种在显微镜引导下的强大切割系统,以紫外光作为接触和无污染的刀,感兴趣的细胞被切割并收集在不同的容器中,以尽量减少空间信息损失。随后,细胞可以通过直接的RNA-seq或预先标记空间条形码的多重过程进行分析。Moor和同事利用LCM和bulk RNA-seq在空间上分析肠绒毛片段,然后在平行scRNA-seq实验中根据共同表达的标志性基因将单个肠细胞定位到其推测的原始位置。LCM能在微米分辨率下精确地从组织中分离出感兴趣的区域,其应用更多集中在单细胞分离。
这种方法通量很低,而且对操作要求很高,但是基于LCM的转录组学或基因组学成功实现单细胞的空间转录组。
2.3 基于图像的原位转录组
基于图像的原位转录组解决了细胞通量的问题。最初的方法称为单分子FISH (single molecule FISH,smFISH),能够检测单个转录本,被设计用于同时检测一个到几个(典型的3或4个) mRNA的亚细胞分辨率FISH。随后,研究者尝试用多重smFISH来增加每个细胞位点的可检测mRNA的种类。扩展FISH(Expansion FISH,ExFISH )被用于数十个培养细胞中探索7个靶基因,而邻近连接原位杂交(PLISH)可测固定组织中8个基因。亚序列的ouroboros单分子FISH(ouroboros singe-molecule FISH,osmFISH )能够同时测定来自组织切片的成千上万个细胞的33个基因。序贯FISH(Sequential FISH,seqFISH )可以通过多轮杂交对单细胞内转录本进行条码化的方法。如框1所示,在这项技术的发展过程中,检测能力从固定细胞中的12个基因增加到组织切片中的10000多个基因。
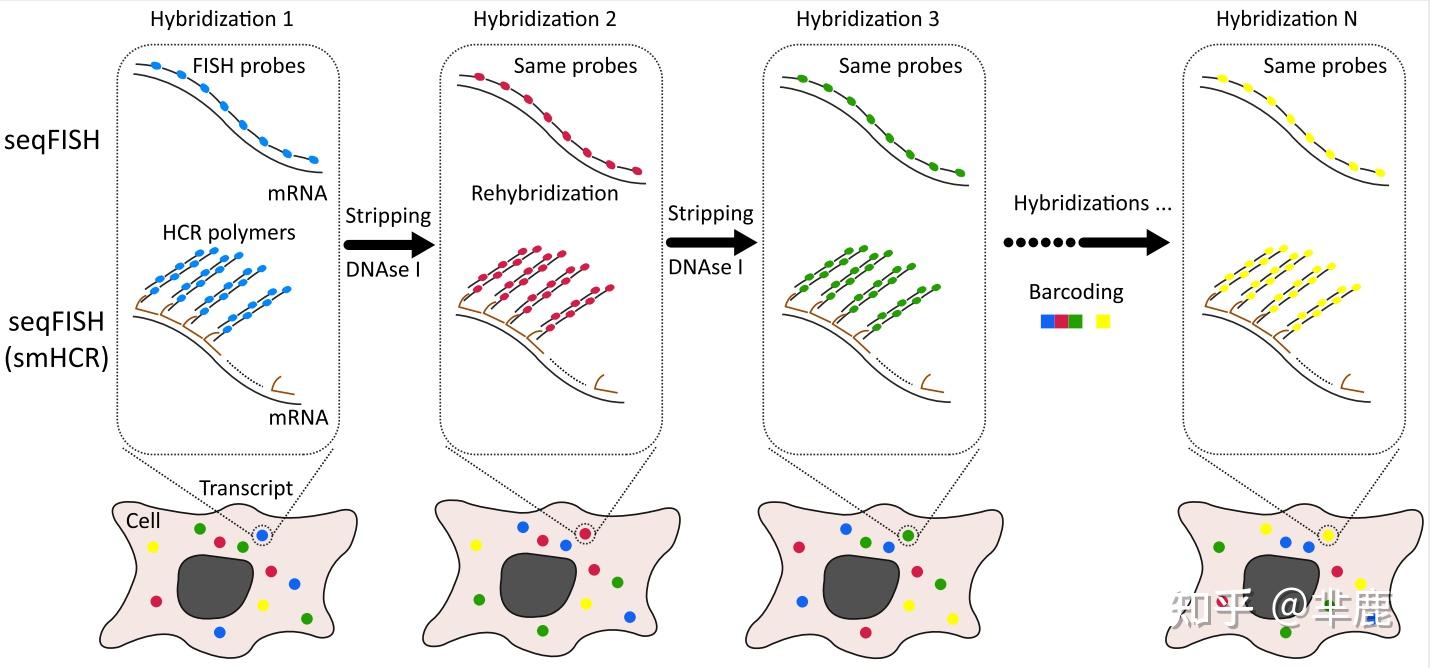
seqFISH和细胞成像示意图
每一轮smFISH中,每个转录本与一组已知颜色荧光标记的探针杂交,成像记录其位置和颜色,然后经DNaseⅠ处理去除。在接下来的一轮中,相同的探针被重新杂交到转录本上,但耦合到不同的荧光团。由于转录本是固定的,在几轮杂交过程中保持不变的点可以标记为exclusive转录本。通过对时间颜色序列的解码,可以识别出原始基因。与smFISH相比,smHCR和seqFISH的结合提高20倍的信噪比。简称:seqFISH,序贯荧光原位杂交,smFISH,单分子荧光原位杂交,smHCR,单分子杂交链式反应。
ISS和FISSEQ的基本理论和图式。ISS的靶向方法可以在固定的细胞或组织中分析靶向基因。mRNA最初被原位反转录成cDNA。与STARmap类似,cDNA随后与条形码锁探针连接,通过连接循环,通过目标引物滚动循环扩增(RCA)扩增。将得到的微米分辨率的滚动循环产品(RCPs)经过多次成像循环后读出条形码进行测序(图I,顶部 )。
相比之下,FISSEQ理论上提供对单个细胞内整个转录组的无偏倚测量,而不是针对特定基因。RNA在固定的细胞中用随机六聚体在5′端带有一个标签(cyan)进行反转录。将制备好的cDNA在RCA前循环,然后用交联剂[BS(PEG)9]将其交联到细胞-蛋白基质固定化。RCPs通过SOLiD测序,读取长度为30个碱基(图I,底部)。最近,新一代FISSEQ(INSTA-seq)已经启动,随着使用NGS组装的阅读框更长,FISSEQ 2(INSTA-seq)检测到以前因短读限制而未解决的小调控变体。
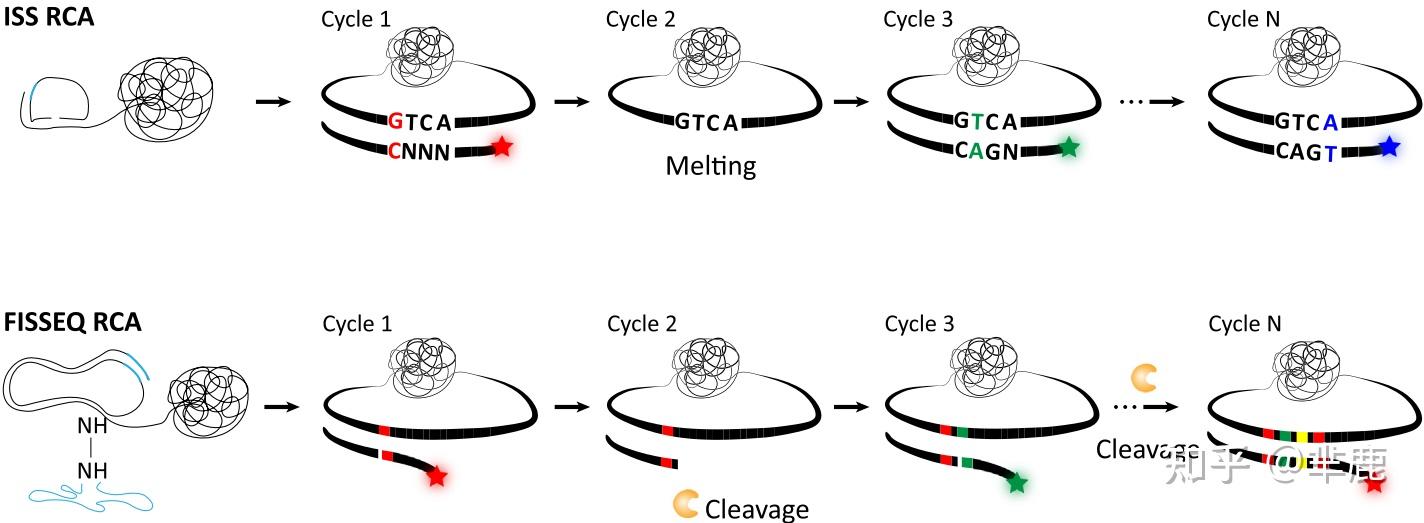
两种ISS方法的示意图
(顶部)cDNA经靶引物RCA循环扩增,形成DNA纳米球。通过连接化学方法对RCPs进行测序,以解码锁探针内的4基条码。(底部)用[BS(PEG)9]交联细胞蛋白质基质,固定由多个cDNA序列串联重复组成的RCA产物,用SOLiD (寡核苷酸连接和检测测序)对短读片段进行无偏测序。缩写:ISS,原位测序,RCA,滚动循环扩增。 2.4 空间编码(Spatial Barcoding)及NGS
利用探针代表RNA物种的靶向原位方法存在不足,越来越多的研究聚焦在相对较小的尺度上直接原位测序或组织切片空间标记。空间转录组学(Spatial transcriptomics,ST)就是其中一种方法,开发带有分散的100μm微孔的定制载玻片来捕获mRNA。每个孔涂上包含空间条形码的寡核苷酸,这是对应孔的专属空间条形码和poly T尾巴,以捕获带有poly A的mRNA。在一个6毫米×6毫米的正方形中,总共产生1007个条形码微孔。微孔内的寡核苷酸可以捕获透化组织切片中的mRNA。cDNA合成在载玻片上进行。随后,使用基于NGS的RNA-seq生成转录组,通过将空间条形码映射回每个微孔,最终解码位置信息。
最近开发的高分辨率空间转录组学(high-definition spatial transcriptomics,HDST)使用分裂池方法生产高分辨率(2μm),高密度(百万)珠阵列。与ST相比,HDST显著提高空间分辨率。然而,HDST平均每个池有7个独特的分子标识符(unique molecular identifier,UMIs),严重限制其测量高丰度基因的能力。
随后开发的Slide-seq给高通量实验提供足够的空间特征。在Slide-seq中,首先将一层10μm独特条形码的微粒(珠子)包裹在橡胶涂层玻璃覆盖面上。与其他高通量scRNA-seq方法中使用的微珠类似,每个微珠上的条形码是随机分布的。因此,在对每个珠状物进行条形码寡核苷酸鉴定之前,必须先将它们放在一起进行测序。
为解决这一问题,Slide-seq利用寡核苷酸连接和检测(sequencing oligonucleotide ligation and detection,SOLiD)化学的测序方法,随后获得每个珠上不同的条形码。在RNA-seq之前,在每个10μm点上解析唯一的空间标识符,寡核苷酸捕获的mRNA的表达可以映射回其原始的空间坐标。因此,Slide-seq以堪比单个细胞大小的分辨率对150万个点进行测序,但Slide-seq与Drop-seq相比,每个珠子的UMIs更少。
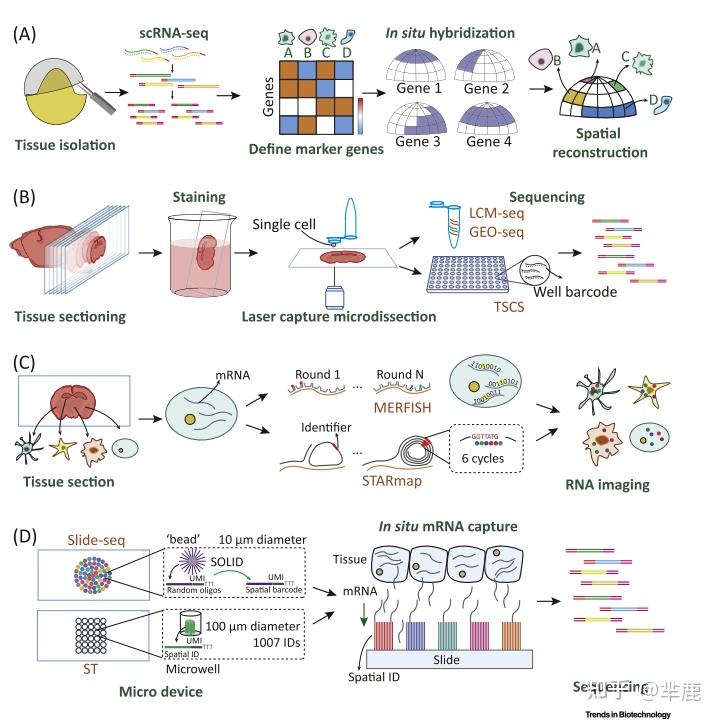
四种空间分辨转录组学的模式图
(A)空间重构的计算机方法。以Seurat为例,斑马鱼胚胎膜首先分离并消化成离散的单细胞。后续scRNA-seq用于确定每个细胞类型的基因标记。采用原位杂交技术对各标记基因的表达分布进行分析。最后,单个细胞可以被定位膜上特定的"仓"内。
(B)基于LCM的方法。冰冻组织切成10μm切片,染色后用特定颜色标记单个细胞。划线细胞由LCM分离,然后在单独的管子(LCM-seq和GEO-seq )或96孔板( TSCS,装载有良好的条形码)中裂解。最终,每个细胞内的转录本都可以被测序。
(C)原位RNA成像方法。探针在切片的冻存组织中转移到每个细胞中。对于MERFISH,N轮荧光读出可以对每个mRNA进行二进制编码,通过解码可以确定mRNA的种类。对于STARmap,6个循环的荧光读出可以解码条码序列,然后定位与信号放大探针杂交的mRNA。
(D)原位测序方法。mRNA被预加载的条形码原位捕获,这些条形码代表空间位置。测序完成后,通过对条码序列的解析,映射出原始地址。简称:GEO-seq:地理位置测序、LCM:激光捕获显微切割、MERFISH:多重抗误差矫正荧光原位杂交技术、scRNA-seq:单细胞RNA测序、SOLiD:寡核苷酸连接检测测序、ST:空间转录组学、STARmap:空间分辨转录放大读出图谱、TSCS:空间单细胞测序、UMI:唯一分子标识符。 3 前沿空间转录组技术的应用
3.1 发现疾病的空间异质性
许多生物过程中基因表达呈现空间模式,很难通过传统的批量基因分析或经典scRNA-seq发现这些模式。在疾病的发生发展过程中,空间异质性具有根本的重要性,了解细胞类型及其在病变组织中的定位有助于解析空间转录组。
3.2 建立空间转录组图谱
单细胞转录组图谱有助于在分子水平上研究生物结构。美国国立卫生研究院(NIH)将在未来7年内投入大量精力开发一个前所未有的人类生物分子图谱。美国国立卫生研究院共同基金人类生物分子图谱计划(HuBMAP)旨在通过支持技术开发、数据采集和详细空间绘图,建立一个全球可访问的框架在单细胞分辨率下全面绘制人体细胞图谱。
3.3 描绘胚胎发育和空间蓝图
胚胎发育是在生物分子水平上发生快速动态变化的复杂过程,对这种微小变化的时间和空间方面的分析_一_直具有挑战性。空间转录组学技术有望解析胚胎发育的空间表达蓝图。
4 结论和展望
空间转录组在发现疾病因素、建立空间图谱和描绘空间蓝图等方面得到广泛的应用和推广,但其潜力远远不止于此。例如,在细胞间相互作用的研究中,从转录组数据和已知的配体-受体复合物推断不同细胞类型相互作用,但单细胞间持续的相互作用很难立即捕获。无论是在组织中还是在培养环境中,封闭空间内的细胞更容易发生相互作用,这正是空间转录组的应用所在,因此有望将其引用到细胞间相互作用的研究中。一些巧妙的实验表明,缺血神经元的体细胞和轴突线粒体在单细胞尺度上是相通的。此外,scRNA-seq技术多方面促进空间转录组的发展,例如从细胞分型中提供标记基因,有助于scRNA-seq通过基因的空间分布来区分亚群。
基于图像的方法如seqFISH+、MERFISH和STARmap提供观察单个细胞内分子行为的亚细胞视图,因此,分析基因-基因交互作用组、基因调控网络和多模态组学是可行的。研究表明,DNA变异与mRNA表达有关,因此,同时检测分子的原始坐标可以加深对转录因子如何激活基因表达、基因如何相互沟通以及细胞如何功能的理解,未来前进的方向是突破这些突出问题的瓶颈。
其中的主要问题是原位获取单细胞的无偏转录组,因为大多数现有的基于实验的方法都来源于靶向方法smFISH。SMR、单分子杂交链式反应(single-molecule hybridization chain reaction,smHCR)和osmFISH具有优异的检测效率和信噪比,但都受到目标数的限制。相比之下,seqFISH+和MERFISH可以原位测量来自众多细胞的10000多个mRNA,但它们不能提供新序列或小序列变异的无偏倚检测。
FISSEQ在有针对性和无偏倚的方法之间起到折中的作用,但这种方法效率低,难以在组织上开展。此外,仅对短读进行排序并不足以准确检测变体或新片段。基于LCM的方法一旦突破细胞通量低的瓶颈,具有巨大的潜力,因为它们可以通过深度测序获得全基因转录组。
克服这一瓶颈包括3个方向:细胞边缘识别和裂解、分离细胞的多重条形码重新定位和提高显微切割效率。HDST和Slide-seq获得区域分辨率为微米量级的无偏倚转录组和位置信息。因此,需要开发与高通量scRNA-seq相符合的高空间分辨率新技术以解决一系列生物学问题。
一知半解
- 仍然看不懂seqFISH 的原理:(后来终于看懂了!)
- 意思就是,首先第一个探针,第一轮杂交,某一个转录本会呈现一种荧光颜色,然后第二轮杂交,随机出现第二种颜色。颜色是随机的。所以每个转录本检测位点就会得到一个由颜色序列组成的,长度为杂交轮次的barcode,而图片就是记录barcode空间位置的信息。
- 针对3.2 也有一些文章调查统计了当前已有的空间转录组图谱,也有空转相关的数据库,后续可以搜索整理一下
一些想法
- 对于下游的主要研究生物学问题的科研人员来说,其实并不太关心各项技术的细微原理和优劣,我们更关心的是哪个技术能够尽快的商业化实现,并在自己的样本中被应用起来,目前由应用价值的主要就是10X Visium它们家的了,所以很多下游的数据分析方法也都放在解卷积SPOT到单个细胞分辨率的程度上。
- 另外Image-based in situ sequencing似乎也很受欢迎,有不少实验室在研究,说不定在人上商业化的也会很快很方便?
- 针对原理的了解,我觉得囫囵吞枣一般或许也差不多,后续如果真的用到某一种类型的数据再去研究也可,后面还是要花力气去研究数据分析。
参考资料:
综述│TRENDS BIOTECHNOL:空间转录组揭示单细胞分辨率下的器官分子结构(国人佳作)
Liao, Jie; Lu, Xiaoyan; Shao, Xin; Zhu, Ling; Fan, Xiaohui (2021): Uncovering an Organ's Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. In Trends in biotechnology 39 (1), pp. 43–58. DOI: 10.1016/j.tibtech.2020.05.006.
末尾啦~
希望获得一个小小的点赞❤和关注
<hr/><hr/>我是芈鹿,欢迎关注!
生物信息学在读博士生,乐于分享自己的科研感悟。
原文地址:https://zhuanlan.zhihu.com/p/429416267 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-5-26 13:47
发表于 2025-5-26 13:47
 提升卡
提升卡


