常用的蛋白灰度分析软件有 Image-Pro Plus ,Quantity One ,Image J/fiji。(还有不太常用的还有Gel-Pro Analyzer、Image Lab等),所有的蛋白灰度分析软件可以在本文的第四部分下载。
不同软件测量值不同,即使相同软件重复测量数值也不一定相同,因此只要测定数值能反应目的蛋白变化趋势,测量值相对偏差不大即可。不同的软件各有千秋,大家可以综合考虑再做选择。
1、Image-Pro Plus
与我们耳熟能详的image J不同,image-Pro plus是一个付费使用的软件。
Image-Pro Plus 综合性能最好,操作简便结果可靠。因为它有强大的 IOD 算法,能准确反应蛋白灰度。但需要靠魔棒或轨迹法选取目的蛋白区域,有一定的主观性,不过蛋白灰度测定本身就是半定量分析(也就是说测量值本身就没有一定标准)。
剔除背景调整参数之后,有 2 种方法选择目的蛋白区域:魔棒和手绘轨迹法
①魔棒:首选方法,客观科学,适合于蛋白条带轮廓清楚且各蛋白条带不融合(若条带弥散,相互融合,则魔棒无法选择单一蛋白条带,这时就要用手绘通过肉眼选择目的蛋白区域)。
②手绘轨迹法:主观性较强,当魔棒无法选择单一条带时才使用。
2、Quantity One
Biorad公司的Quantiy One软件,付费软件。其最大的优点莫过于自动识别条带代替人工分析以降低主观误差,同时它的很多功能渗透了艰深的数理理论以及概率统计的原理。
有泳道-轨迹测定法,等高线/手绘选取目的区域测定法等方法。
①泳道-轨迹测定法:先剔除一系列背景,再选择泳道(lane),然后分析条带(band),最后使用高斯建模得出灰度分析值(Gauss Model trace)。它最大优点在于可以完全抛弃人为主观因素进行全自动定量,比较科学。重复性好,但这种方法使用起来步骤较为繁琐。
②等高线-手绘选取目的区域测定法:通过半自动描绘电泳条带的等高线边缘来得到等高线区域内部面积,再将该面积乘以区域内平均光密度值得到条带内部总的信号量。这个方法最简便,但重复性不太好,且等高线的绘制处于“半自动”状态,即需要人为判断作为等高线标准的电泳条带的边缘。
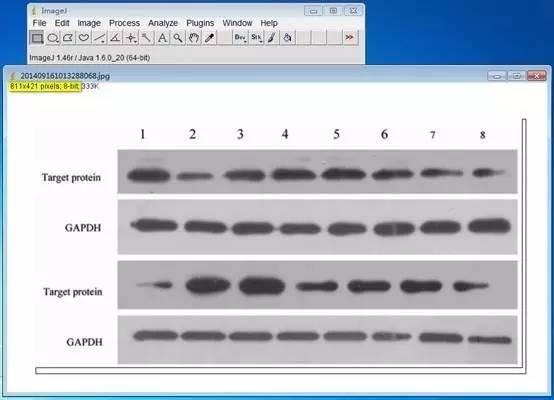
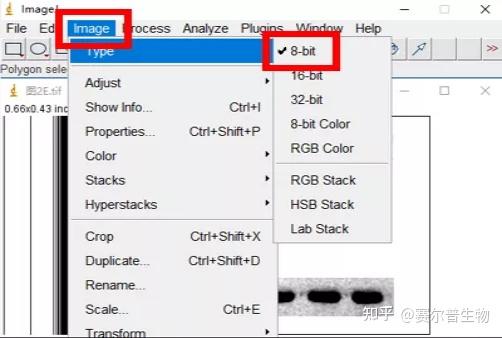
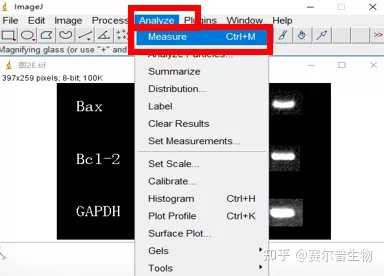
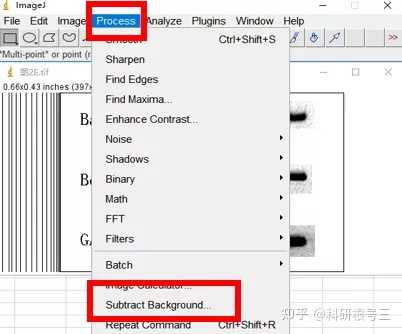
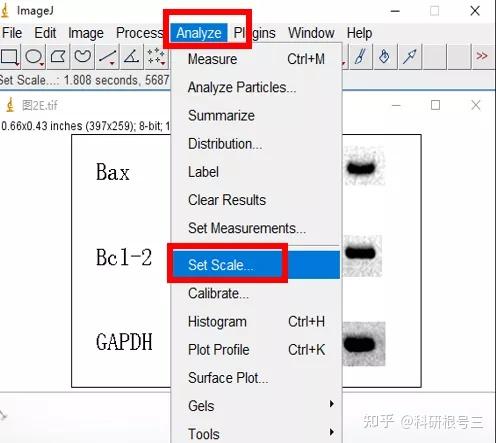
3、Image J & fiji( fiji is just ImageJ)
Image J 这款工具胜在免费,基于强大的图片分析与处理功能,Image J在科研中的应用极为广泛。(使用广泛也就意味着很多人都在用,遇到问题可以更快的找到解答)
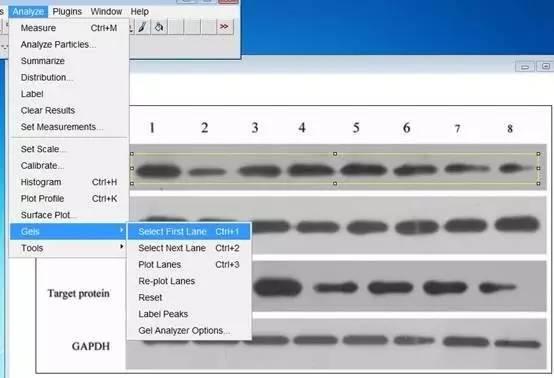
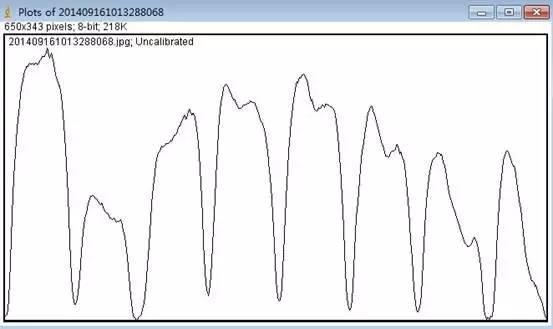
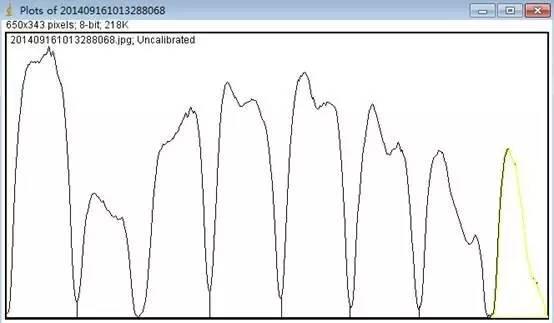
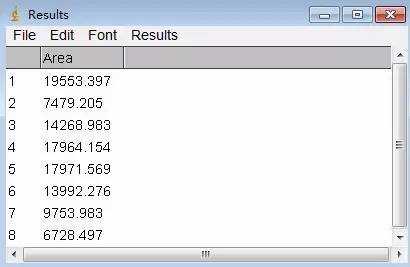
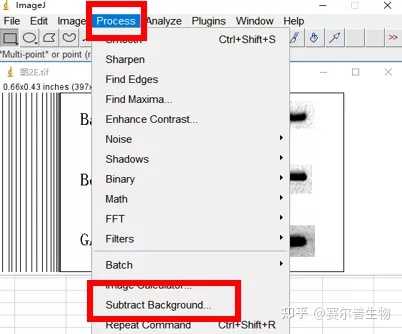
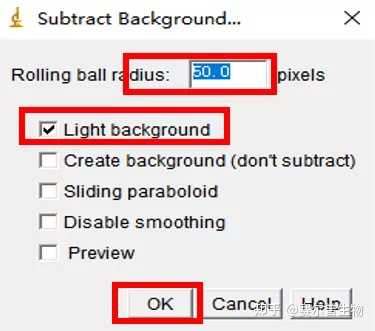
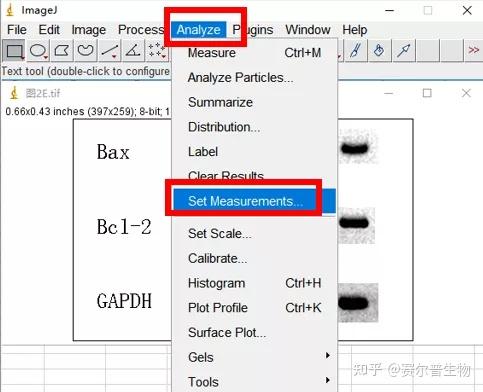
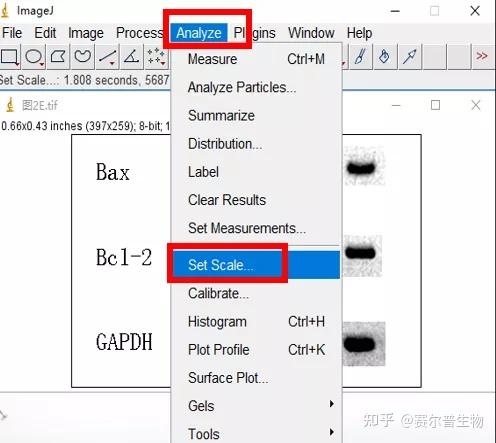
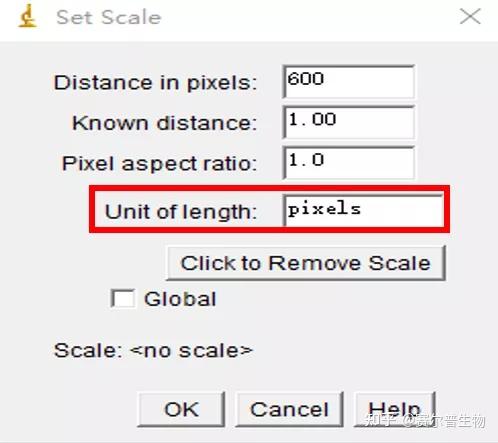
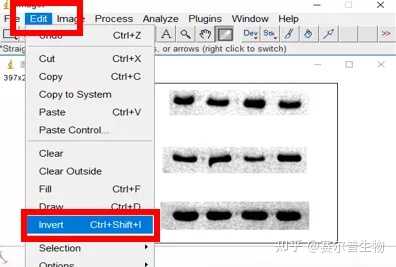
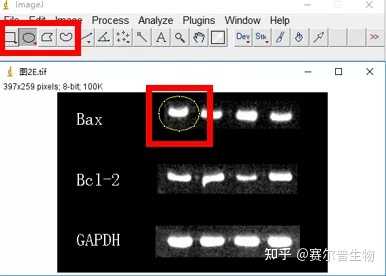
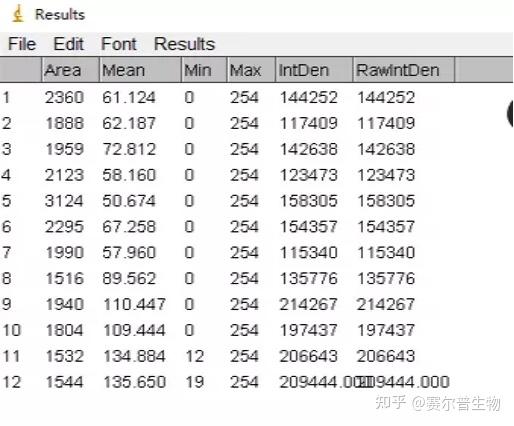
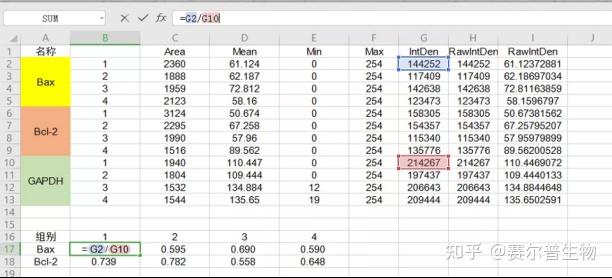
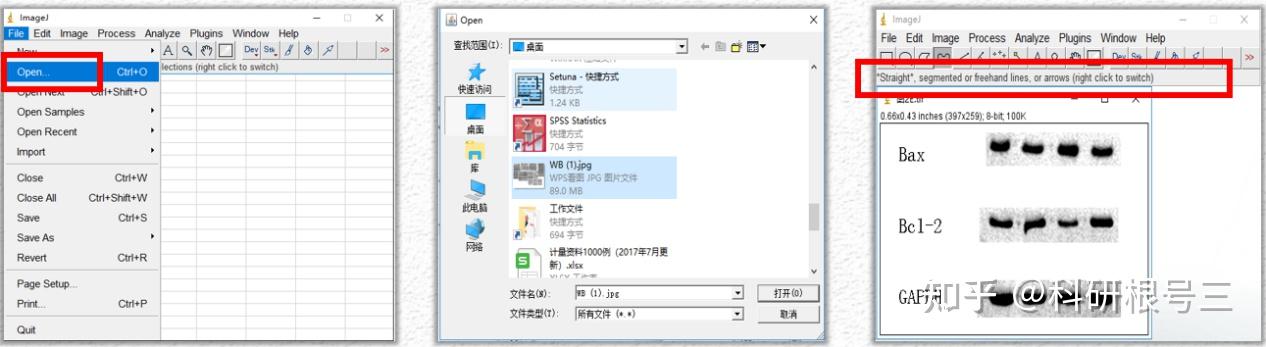
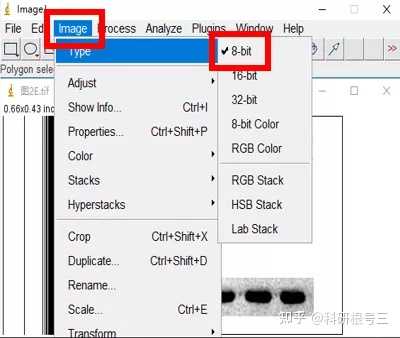
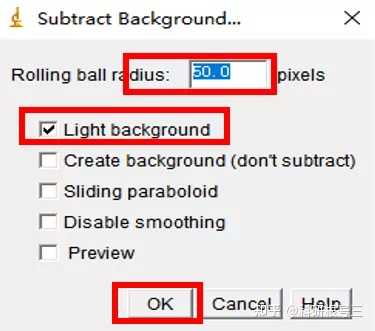
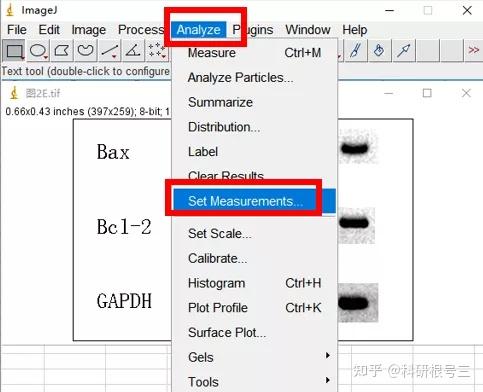
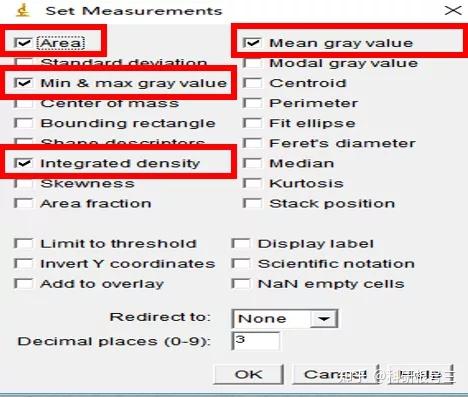
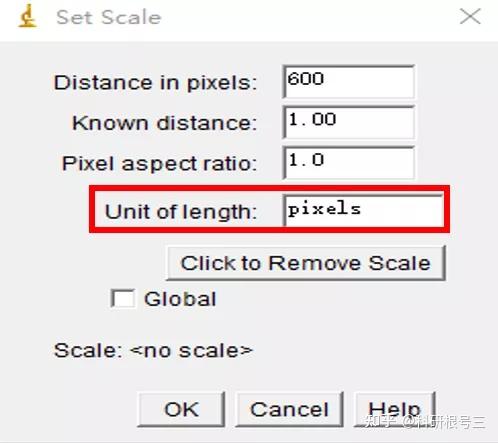
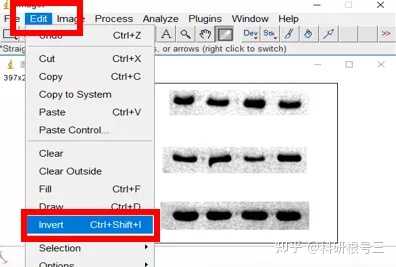
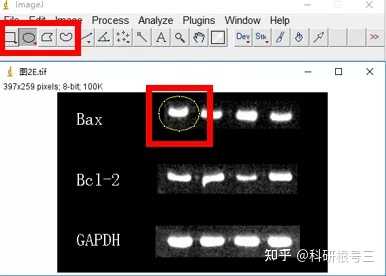
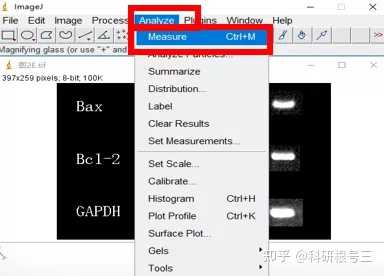
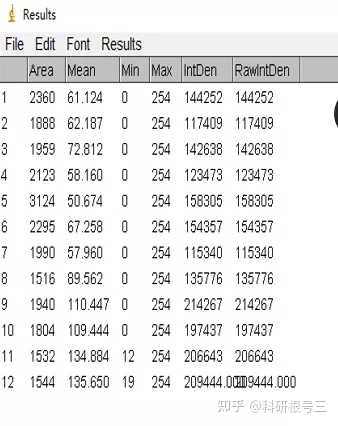
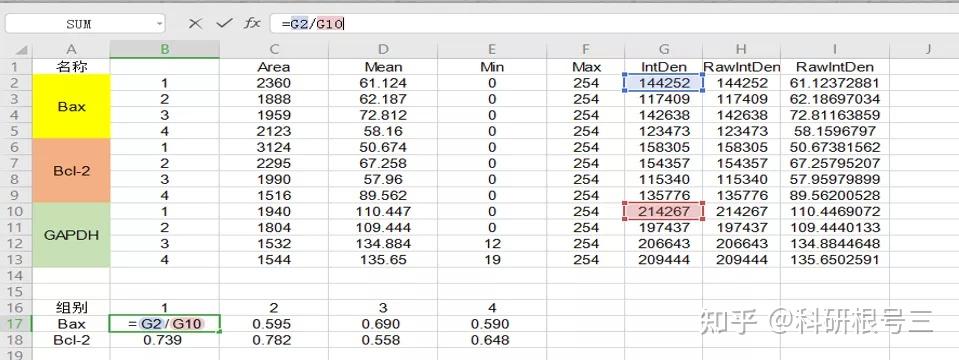
Image J做蛋白灰度分析的步骤操作简单,容易上手。先转化灰度图以及去除背景影响,然后设置参数与单位,转化为亮带之后,利用选择工具选中分析的单个条带分析,之后逐一选取分析即可。
之前分享过详细的操作步骤,需要学习的可以看以下文章: Image J 定量分析 WB 灰度值 (qq.com)
fiji 是 ImageJ 的版本之一,全称为 fiji is just ImageJ,也是免费工具。fiji 就是预装了很多生物医学分析常用插件的ImageJ。
Fiji 做蛋白灰度分析的步骤和Image J 一致,这里不再赘述。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-3-29 19:35
发表于 2025-3-29 19:35


 提升卡
提升卡




 发表于 2025-3-29 19:37
发表于 2025-3-29 19:37