SAM 文件还包含了一些额外的标志位和标签字段,用于描述特定的比对特性和附加的信息。这使得 SAM 格式非常灵活,可以记录各种类型的比对结果,支持多样的分析需求。
需要注意的是,SAM 格式是一个文本文件格式,相对于二进制的 BAM 格式,其文件大小通常较大。在实际使用中,BAM 格式更常见,因为它将 SAM 格式进行了压缩和二进制化,提供了更高的存储效率和读写速度。
SAM 文件可以通过多种生物信息学工具进行创建、转换和处理,如Samtools、Picard、GATK等。这些工具提供了丰富的功能,如排序、索引、筛选、变异检测和可视化等,以支持基因组比对数据的分析和解释。
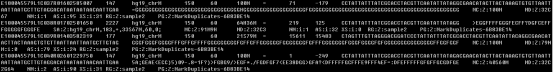
SAM 格式中的每一列代表着不同的含义。以下是 SAM 格式中列的含义:
QNAME:测序读取(read)的名称或标识符。
FLAG:比对标志位,用于描述比对的各种特性和属性。
RNAME:参考序列的名称或标识符。
POS:比对在参考序列上的起始位置。
MAPQ:比对质量分数,表示比对的可信度。
CIGAR:比对的 CIGAR 字符串,描述了比对的碱基操作、插入和删除情况。M:alignment match,表示read比对上或者未比对上(错配);I:insertion to the reference,表示read的碱基序列相对于第三列参考序列有碱基的插入;D:deletion from the reference,表示read的碱基序列相对于第三列的参考序列有碱基的删除;N:skipped region from the reference,表示跳过参考序列的碱基。常出现在转录组数据比对基因组的结果中,N出现的位置表示可能是内含子位置;S:soft clipping (clipped sequences present in SEQ),软裁剪,指read跳过的长度才能比对上;H:硬裁剪,指参考基因组序列跳过的长度才能比对上;比如51M1106N30M,前51个碱基可以匹配,然后跳过参考基因组上1106个碱基后又有30个碱基匹配。
RNEXT:下一个比对的参考序列的名称或标识符。
PNEXT:下一个比对的起始位置。
TLEN:模板长度,表示测序读取在参考序列上的覆盖长度。
SEQ:测序读取的碱基序列。
QUAL:碱基序列的质量分数。
除了这些必需的列,SAM 格式还可以包含一些可选的标签(Tag),以提供额外的元数据信息,如比对算法使用的特定参数、分析结果等。这些标签以格式为 "TAG:VALUE" 的形式出现在每行的列中,例如 "AS:i:100" 表示比对得分为 100。部分标签信息如下:

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-2-20 18:27
发表于 2025-2-20 18:27