金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
非医疗器械方面的质量人员,随便说说。
《医疗器械质量管理体系用于法规的要求》强调的是满足法律法规的要求。该标准在总则中说:“本标准的主要目的是便于实施经协调的质量管理体系的法规要求。因此,本标准包含了一些医疗器械的专用要求,删减了ISO9001中不适于作为法规要求的某些要求,由于这些删减,ISO13485就不是ISO9001标准在医疗器械行业中的实施指南,两者不能兼容。
根据医疗器械的行业特点,ISO13485:2003标准中作了许多专业性规定,如4.2.4记录控制中规定:“组织保存记录的期限应至少相当于组织所规定的医疗器械的寿命期,但从组织放行产品的日期起不少于2年,或按相关法规要求规定。” 6.4工作环境中,增加了对产品清洁、防止污染、人员健康等方面的要求;7.2.3顾客沟通中增加“忠告性通知”;8.2.1的标题改为“反馈”,而不是ISO 9001的8.2.1的顾客满意,并增加了提供质量问题早期报警和评审生产后阶段的经验等内容。因为顾客满意和顾客感知不适宜法规中作为要求来实施。此外对有源植入性医疗器械和植入性医疗器械还有专用要求,即“组织应记录检验和试验人员的身份。”
新的ISO13485标准是一份独立的标准,其章节结构虽与ISO9001:2000相同,某些章节内容也与ISO9001相同,但由于ISO13485标准根据医疗器械行业的特点,突出了法律法规要求,淡化了顾客满意,删减了ISO9001:2000的一些重要要求,因此满足ISO13485的要求,不等于同时满足ISO 9001:2000的要求。 管理方面:建立相应的组织机构,规定各机构的职责、权限,明确质量管理职能,生产管理部门和质量管理部门负责人不得互相兼任;应当建立质量管理体系并形成文件,质量管理体系形成的文件应当包括质量方针和质量目标、质量手册、程序文件、技术文件、作业指导书和记录,以及法规要求的其他文件;
开发方面:建立设计控制程序并形成文件,对医疗器械的设计和开发过程实施策划和控制;
质量方面:建立监视和测量控制程序并形成文件,确定所需要的监视和测量活动,配置相应的装置,对监视和测量装置进行控制;还要建立不合格品控制程序并形成文件,规定对不合格品进行控制的部门和人员的职责与权限,对不合格品进行标识、记录、隔离、评审,根据评审结果,对不合格品采取相应的处置方法;按照医疗器械不良事件监测和再评价管理的要求建立不良事件监测程序并形成文件,明确不良事件管理人员职责,规定不良事件收集方法、报告原则、上报程序和时限;建立数据分析程序并形成文件,规定收集与产品质量、不良事件和质量管理体系运行有关的数据,包括反馈、产品质量、市场信息及供方情况等等。
生产方面:策划并在受控条件下实施所有生产过程编制生产工艺规程、作业指导书等,并明确关键工序和特殊过程;使用适宜的生产设备、工艺装备、监视和测量装置,并确保其得到控制;建立和保持每批产品的生产记录,生产记录应当满足医疗器械可追溯性要求,并标明生产数量和入库数量。
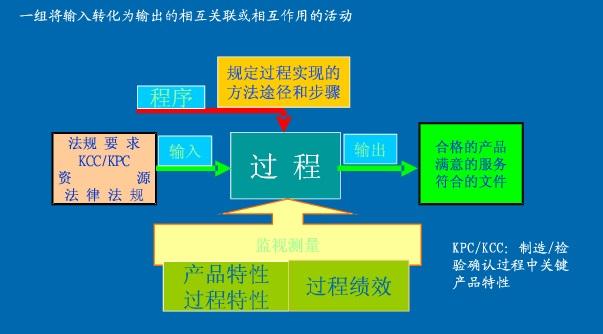
简单来说根据你们公司现在的规模、结构来编写质量管理体系中的质量手册,把质量手册(包含了:适用范围、引用标准、术语定义、管理职责、资源管理、产品实现、测量、分析和改进)写完你绝对入门了。你要的流程也就是过程方法吧,大致的见下图:
 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-10-23 21:15
发表于 2024-10-23 21:15


 提升卡
提升卡 发表于 2024-10-23 21:16
发表于 2024-10-23 21:16





