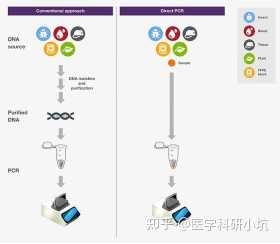
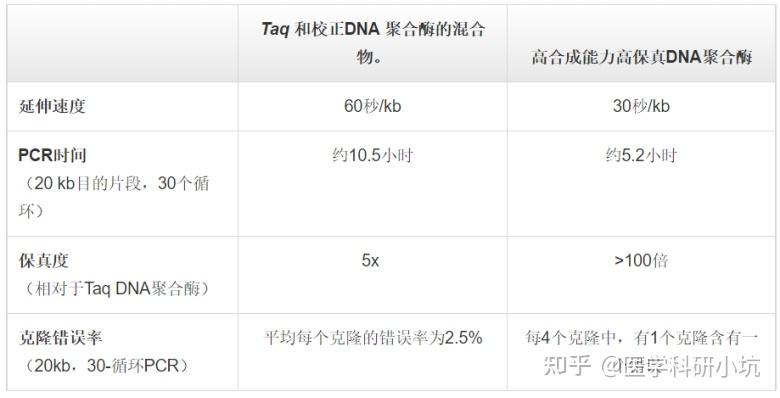
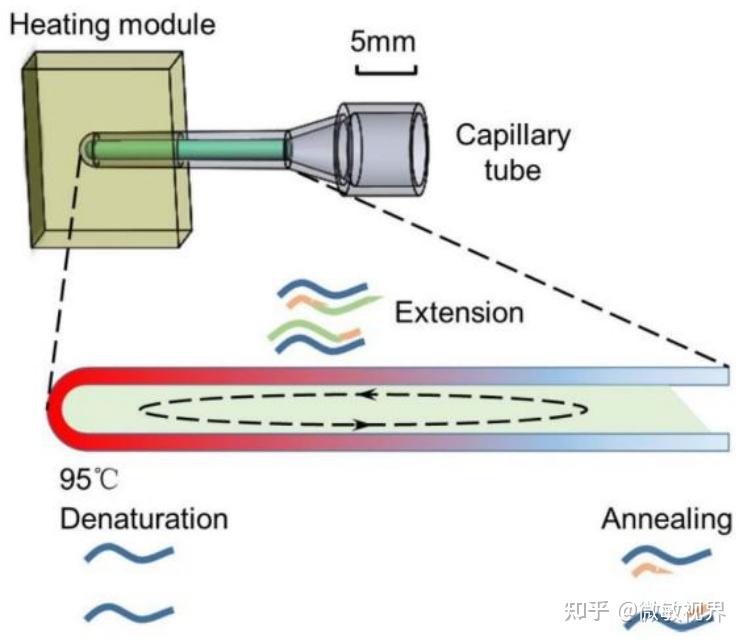
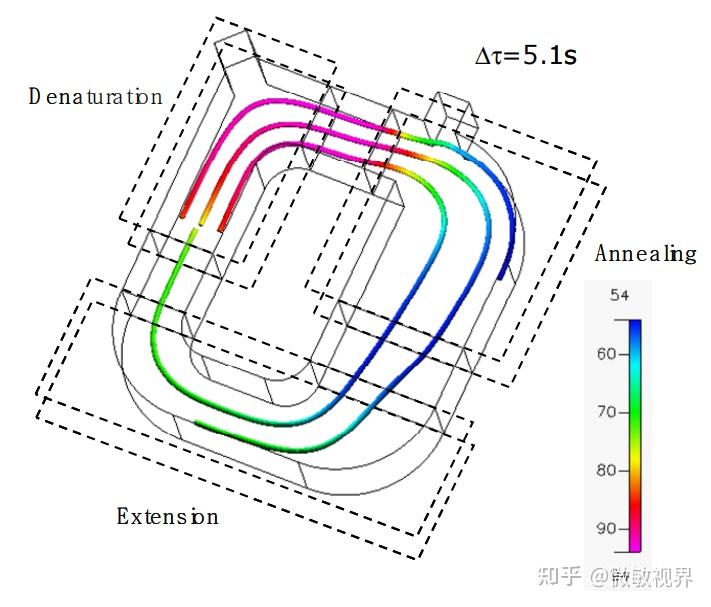
金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
准备好了没有,史上最全PCR知识来了:
PCR 技术
聚合酶链式反应(Polymerase chain reaction)也就是我们通常所称的 PCR 反应,自 1971 年发明以来,已经在医学、微生物学、植物学和动物学等领域得到广泛的应用,特别是 2020 年新冠疫情以来,采用核酸筛查的方法寻找新冠病毒感染人群,可谓是一种快速、适用于大 规模人群的筛查手段,同时被当成阳性病例确诊手段之一,自此 PCR 反应被广大的普通民 众所了解和熟知。众所周知,PCR 反应是测序、分子克隆技术的基础,PCR 技术的发明将 分子生物学技术向前推进了一大步。
生命科学是解决人类重大生命问题的科学,PCR 技术的诞生成为生命科学的生长点, 使生命科学在自然科学中的位置起了革命性的变化。20 世纪 50 年代,遗传物质脱氧核糖核 酸(DNA,Deoxyribonucleicacid)双螺旋结构的发现,开创了从分子水平研究生命活动的新 纪元。此后,遗传信息由 DNA 通过 RNA(Ribonucleic acid,核糖核酸)传向蛋白质这一“中 心法则”的确立以及遗传密码的破译,为基因工程的诞生提供了理论基础。
随着 PCR 技术的发展,对 PCR 技术的使用从测序和分子克隆,逐渐应用于病毒、细菌、 真菌和物种的鉴别和鉴定。甚至,我国的食品安全国家标准(方法标准)中也引入了 PCR 技术。PCR 技术具有高灵敏性、快速和高通量的特点,目前在疾病诊断、致病微生物鉴定、 物种鉴别等领域得到推广和应用。
PCR 技术发展历程
1953 年 4 月 25 日,沃森(James Dewey Watson)和克里克(Francis Harry Compton Crick) 在《Nature》发表 DNA 双螺旋结构模型,自此开创了分子生物学时代,这一发现也被誉为 20 世纪以来生物学方面最伟大的发现,使生命科学的研究深入到分子层次。从此以后,科 学家一直在探索研究 DNA 的新方法,新技术。1971 年,印裔生物学家科拉纳(Har Gobind Khorana)提出“DNA 体外合成”的设想,即经过 DNA 变性,与合适引物杂交,利用 DNA 聚合酶延伸,不断重复该过程便可克隆 tRNA 基因。但是当时引物合成技术水平有限,尚未 发现较好稳定性的 DNA 聚合酶,所以科拉纳的设想逐渐被遗忘,当时 DNA 体外扩增技术 并未获得全面推广。1973 年,我国台湾科学家钱嘉韵从黄石公园热泉中的嗜热细菌,海栖 热孢菌(Thermus aquaticus)中分离出耐高温的 Taq DNA 聚合酶,并于 1976 年发表于《细 菌学杂志》(J. Bacteriol)。
第一代传统 PCR 技术
1983 年 4 月的一个星期五的晚上,美国化学家穆利斯(Kary Banks Mullis)驾车在高速 公路上飞驰,脑海中猛然闪现出 DNA 双链的结构,以及让 DNA 片段不断自我复制的想法。 1984 年 11 月,穆利斯对一 49 个碱基对(bp,base pair)的 DNA 片段进行了 10 个 PCR 循 环的复制扩增,成功完成了第一次 PCR 实验。1985 年,穆利斯阐述了 PCR 技术的基本原理, 即在试管中模拟细胞内 DNA 复制,具体包括提供 DNA 体外合成合适的条件,即模板 DNA、 寡核苷酸引物、4 种核苷酸(dNTP,deoxy-ribonucleoside triphosphate)、DNA 聚合酶,合适 的缓冲液体系,通过 DNA 变性、复性及延伸的温度与时间。最初,DNA 聚合酶使用的是 大肠杆菌 DNA 聚合酶,该聚合酶不耐热,每次加热变性 DNA 后都要重新补加。1986 年, 穆利斯将钱嘉韵发现的耐高温 Taq DNA 聚合酶应用于 PCR 反应,极大地减化了 PCR 工作 流程。
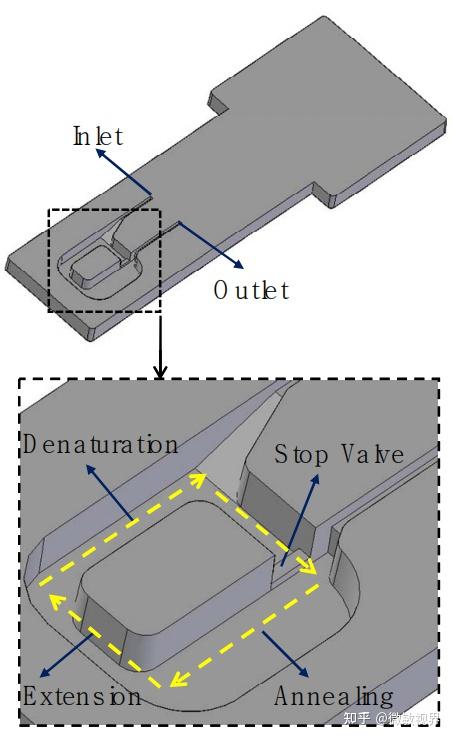
当时,PCR 实验仍需纯手工操作:设置 3 个不同温度的水浴槽,然后按照高温变性、 退火、延伸的顺序,将 PCR 管浸泡在不同温度水浴槽中,完成 DNA 变性、引物结合和 DNA 合成的 PCR 循环,完成一次 PCR 实验需要几十个循环,十分费时费力。1988 年穆利斯所在 的西特斯(PE-Centus)公司发明了第一台 PCR 自动化循环仪,在耐高温 Taq DNA 聚合酶的 配合下,实现了 PCR 技术的实际应用。1989 年,美国《Science》杂志将 PCR 列为十余项
重大科学发明之首,并将 Taq DNA 聚合酶命名为“年度分子”。1993 年,穆利斯因 PCR 技术被授予诺贝尔化学奖。
第二代定量 PCR 技术
20 世纪 90 年代早期,罗氏(Roche)的科学家团队率先在 PCR 反应体系中加入溴化乙 二胺,将样品置于紫外光下检测 PCR 终点荧光信号,可实现目标基因的定性检测。但这仍 然给科学家们留下了一个问题——如何更容易地量化扩增产物的数量,从而确定他们开始扩 增的 DNA 的数量。定量 PCR(Quantitative PCR, qPCR)的概念被提出来,即在在 PCR 反 应体系中加入荧光化学物质,随着 PCR 反应的进行,PCR 反应产物不断累积,荧光信号强 度也随之等比例增加,每经过一个循环,报告一个荧光强度信号,当报告荧光信号的数量达 到一定的阈值时,可以测量相对数量的 DNA (PCR 产物)——越早达到阈值,样本的初始量 越多。这种连续的荧光测量是当今 qPCR 技术的基础,又被称为实时荧光定量 PCR(Real-time fluoroscence quantitative PCR, RT-PCR)。
初始的 RT-PCR 通过在 PCR 反应中使用能够与 DNA 结合的荧光染料,如 SYBR GREEN 染料,来实现对 PCR 过程的实时检测。SYBR GREEN 与双链 DNA(dsDNA)结合力远高 于溴化乙二胺,结合后能够释放很强的荧光信号,提高了 PCR 检测灵敏度,缩短了 PCR 实 验周期。由于 SYBR GREEN 释放的荧光信号强度与 PCR 产物数量成正比,可通过 Ct 值和 标准曲线对样品中的 DNA(或 cDNA)的起始浓度进行准确定量。20 多年来,以 SYBR GREEN 等荧光染料为基础的 RT-PCR 方法仍然是基础科学研究中最流行的选择之一,但仍 有一些问题,包括产物特异性和扩增目标的信号的不确定性。DNA 结合染料与所有双链 DNA 结合,无法区分非特异性引物退火或伪引物二聚体所产生的多个产品。为了弥补荧光染料的 缺点,在 PCR 反应中,引入了第三个寡核苷酸序列,即探针(Probe),其上面标记了荧光 修饰基团,与目标序列特异性结合后,可在特定波长的激发光下释放出荧光信号,信号强度 与目标 DNA 成正比。由于探针只有与特异性序列结合后才能被激发出荧光信号,与 DNA 荧光染料相比,提高了荧光信号释放的特异性。
2000 年日本学者 Notomi 在《Nucleic Acids Res.》杂志上公开了环介导等温扩增技术 (Loop-mediated isothermal amplification,LAMP)。2009 年,日本荣研化学株式会社(以下 简称“荣研公司”)研制出 H1N1 环介导等温扩增法检测试剂盒,可通过早期快速诊断对防止 该病症的快速蔓延起到积极作用。目前,LAMP 技术具有高特异性、高灵敏度,操作简单、 对仪器设备要求低等优势,扩增结果可通过观察白色浑浊或绿色荧光的生成来判断,已被广泛应用于病毒、细菌、寄生虫等引起的疾病检测、食品化妆品安全检查及进出口快速检测。
第三代数字 PCR 技术
1999 年肿瘤基因组学专家福格尔斯泰因(Bert Vogelstein)和金兹勒(Kenneth W Kinzler) 首次在美国科学院院刊 PNAS 上提出“第三代 PCR”数字 PCR(Digital PCR, dPCR)的概念, 通过将样本分充分稀释,分配到不同的 PCR 反应单元,每个单元包含≤1 个拷贝的 DNA/RNA 模板,每个反应单元内均单独进行 PCR 扩增反应,扩增结束后对各个反应单元的荧光信号 进行统计学分析,以实现 DNA/RNA 绝对定量及稀有等位基因的检测。福格尔斯泰因和金 兹勒将该项技术应用于研究肿瘤罕见突变研究,并于 2003 年进一步提出基于小珠(Bead)、 乳浊液(Emulsion)、扩增(Amplification)、磁性(Magnetic)的 BEAMing 技术,将 dPCR 技术和流式技术进行结合,应用乳状液实现单个试管中实现 PCR 反应体系的分隔。
随着微流控芯片、油包水乳化液滴和纳米制造技术的发展, dPCR 的基本方法已经逐 渐建立。根据反应单元的不同分隔形式,形成了微反应室/孔板 dPCR(Chamber Digital PCR, cdPCR)、微流体 dPCR(Microfluidic Digital PCR,mdPCR)(大规模集成微流控芯片)和微 滴式 dPCR(Droplet DigitalPCR,ddPCR)三大体系。2006 年,Fluidigm 公司推出了第一台 商业化的基于芯片的商品化数字 PCR 系统——IFC 平台。该平台使用物理矩阵的策略,可 将 48 个样品逐个分布在 770 个微反应单元中。2013 年,Life technologies 推出 QuantStudio3D 数字 PCR 系统,采用高密度纳升流控芯片技术,将样本均匀分配至 20000 个单独的反应孔 中。BioRad 公司采用油包水乳化微滴技术,借助微滴发生器,将样品均分成数万个微液滴。 截至目前,美国 Thermo-Fisher 的 QuantStudio 3D 系统(cdPCR)和 Bio-Rad 的 QX200 系统 (ddPCR)占据了 dPCR 的绝大多数市场份额。
此外还有
各种PCR技术的介绍:
实时荧光PCR环介导等温扩增(LAMP)数字PCR原位PCR以及
最新PCR技术进展
其他PCR技术进展此外我还整理了
PCR实验室其他方面的知识
DNA(RNA)模板的提取和准备PCR仪校准溯源PCR 实验室的建设及质控常见问题及解答等等内容。
实在太多放不下,大家可以点个关注加个收藏慢慢品尝~~~ |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-8 12:28
发表于 2024-9-8 12:28


 发表于 2024-9-8 12:28
发表于 2024-9-8 12:28