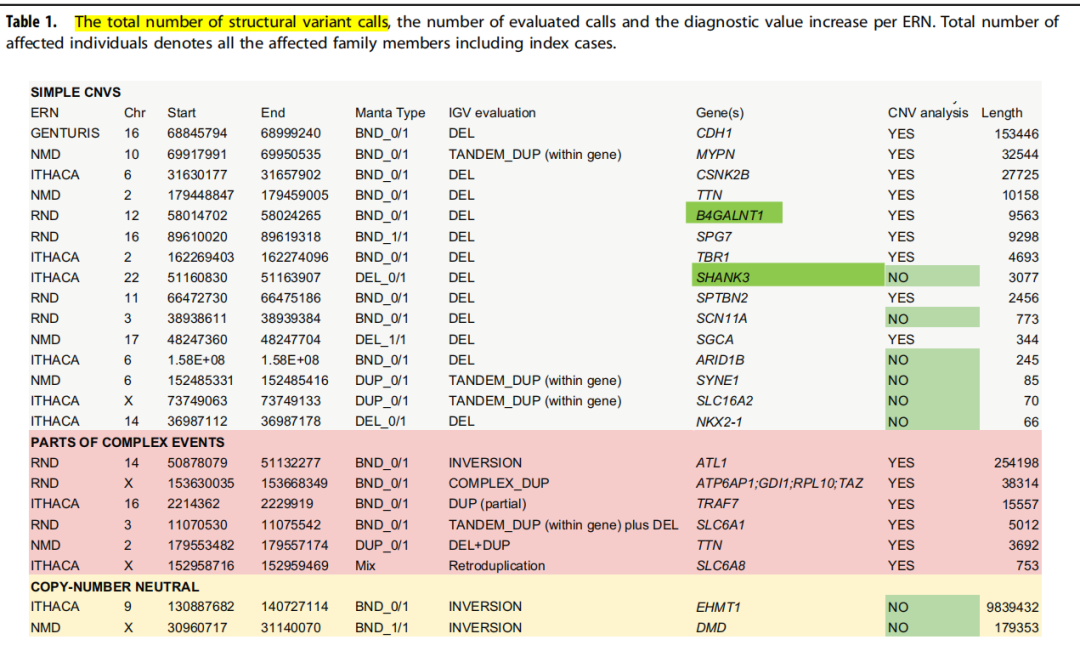
基因组结构变异(structural variations, SVs)通常指基因组的序列和位置变化,如>50 bp的插入或删除(indel)、串联重复(tandem repeat)、染色体倒位(inversion)、染色体内部或染色体之间的易位(translocation)、拷贝数变异(copy number variations, CNVs)及其他形式更为复杂的变异。 由于SVs的断裂点更有可能发生在人类基因组的>98%非外显子区域,因此使用WES数据很难检测到SVs。 读长匹配策略(Read-pair, RP)和读长分割比对策略(Split-read, SR)仅限于ES的读长覆盖区域的断点分析。因此,WES数据主要局限于使用读长深度(Read-depth, RD)策略分析CNVs(缺失和重复)。 然而,WES数据中的RD信号会受到一些因素影响,例如GC含量,因此无法可靠地检测小的CNVs(涉及一个到几个外显子),如果再加上使用RP和SR分析,小的CNVs能更容易被检测到。 尽管WES分析SVs存在困难,但大约2%~4%的SVs(取决于外显子组试剂盒目标区域的大小),预计断点会发生在足够靠近目标区域的地方,使其能够被检测到。 研究对象及方法6,224例患病个体的WES数据(来自5,825个家庭和3,090名未患病亲属),均来自“Solve-RD——解决未解决的罕见疾病”研究计划("Solve-RD solving the unsolved rare diseases"),这是欧盟委员会资助的一项为期五年(2018-2022)的研究项目。 使用BWA-MEM 0.7.8软件将Reads与GRCh37 hs37d5人类参考基因组进行比对。 基于读长匹配策略(Read-pair, RP)和读长分割比对策略(Split-read, SR),利用WES数据,使用Manta SV caller软件,分析SVs。 除了Manta,作者还测试了InDelible,这是一款专门为WES数据开发的用于分析SVs。(虽然在队列中应用indeleble,没有发现额外的SVs) 研究结果在6,224例患者中,检出32例(0.51%)有SVs,涉及23个家系的23种不同的SVs(被认为是致病的)。
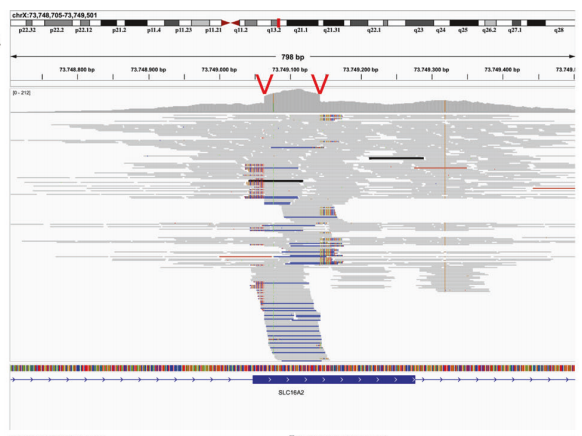
在这23个SVs中,基于RD策略的CNV分析方法(使用了ClinCNV、ExomeDepth、Conifer)未检出8个(0.13%的受检者)(上图中的绿色部分)。 为了与基于RD的CNV分析方法比较,将识别出的SVs分为三类: ①简单的缺失或重复(CNVs)。15个SVs归类为简单的CNVs,其中5个由于长度太小无法通过CNV方法检测到(涉及单个外显子或单个外显子的一部分,大小范围66~3077个碱基)。在这种情况下,只能依赖基于RP和SR进行CNV分析,因为基于RD分析CNV时,覆盖范围在标准噪声水平之外没有明显变化。 最长的CNV是涉及3077个碱基的缺失(无法通过基于RD分析CNV),仅涉及SHANK3基因 (NM_001372044.1) 的第23号外显子的36个碱基,该外显子总长度为2.2 kb。 在基于RD的CNV分析中,由于样本中存在过多的候选CNVs,未发现影响隐性基因B4GALNT1的一个简单缺失(9563个碱基缺失),但是经过优化,减少了CNVs的信号,最终能够被发现。同时也发现了,另一个等位基因上的错义变异,因此构成复合杂合,能够解释患者的表型。
基于RD的CNV分析方法漏掉了70个碱基的简单重复
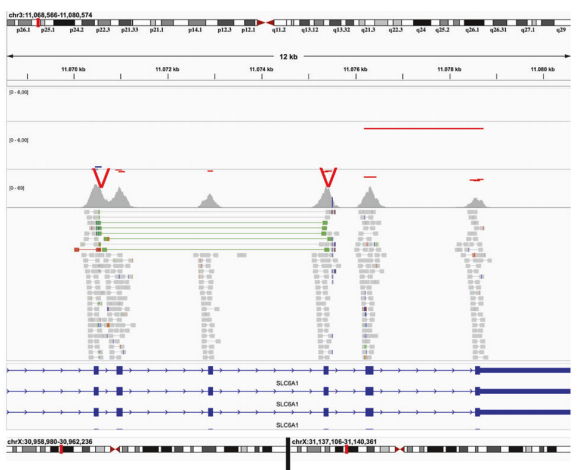
一个复杂的SV:配对读长的距离表示串联重复的存在。基于RD的CNV分析发现在检测到的重复序列旁边存在一个缺失,两者共同形成一个复杂的SV。 ②复杂的SVs(缺失、重复、倒位的组合),其中一部分可以通过基于RD的CNV分析检测到。在这种情况下,使用ES数据几乎不可能检测到所有断点,但可以提示基因组可能存在复杂的SVs。(上图种橙色部分) 在这种情况,RD策略需要联合RP或SR策略进行分析。 ③中性拷贝数变异。检出2例致病性倒位,这两例倒位均发生于表型非常符合的患者。Manta并没有将变异类型报告为INV(倒位),而是BND(断点),因此,为了识别倒位,必须可视化地核查所有断点。 第1例:涉及近10Mb的倒位(g:9:130,887,682-140,727,115, hg19),影响EHMT1基因第25内含子。该患者符合Kleefstra综合征1型(Kleefstra syndrome type 1, KS1)的诊断标准,KS1是一种以精神运动发育迟缓、认知障碍、行为障碍、面部畸形、颅骨形状异常、手异常和先天性心脏缺陷为特征的综合征型神经发育疾病。考虑到提示的异常表型,该患者之前已经使用过阵列比较基因组杂交(aCGH)和EHMT1基因的Sanger测序。由于并没有在候选基因中检测到任何可能的致病性变异,导至患者的诊断延迟了几年,尽管有强烈的临床表型提示。400条带分辨率的核型分析没有该倒位,但通过FISH验证存在倒位。
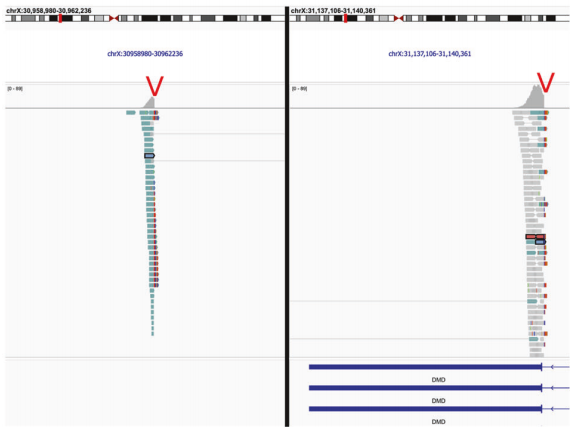
第2例:倒位片段大小约为180kb,影响DMD基因。
综 上 利用WES数据进行重分析,可以从基于RP或SR的SVs分析方法中获益。 在本研究中,大多数CNVs(65%, 15/23)是可以通过基于RD的CNV分析方法发现的。 分析WES数据时,成功识别SVs的机会与SVs的长度无关,但断点必须在外显子区域,且能被大量的具有异常的方向或插入片段长度(insert size)的读长所覆盖。 在WES数据中检测到的CNVs不能像基于SVs分析法那样提供准确的断点信息,因此,在ES数据中,使用基于RD或SR的SVs分析法可以提供有价值的附加信息。 作者没有对WES中的SVs分析策略的敏感性进行评估,因为敏感性很低。 使用Manta或类似工具进行SVs分析不能取代基于RD的CNV分析,因此只能作为补充手段,仅带来微小的获益。 总的来说,利用WES数据发现基因组结构变异还是相当困难,也需要非常仔细地挖掘数据,也相当“考验”外显子组试剂盒的设计方案,总体上利用WES分析SVs获益较低,但也存在可能性。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号