搭建一个可以真正的实际应用的数据库,需要花费很大的精力,主要的难点是,保证全的基础上,也要保证得到的序列是正确的,如果有错误的序列掺入其中,会给后续分析带来很多错误和问题,因此,保证正确是非常必要的。我今天简单以Streptococcus pneumoniae为例,搭建一个简易的数据库,进行引物特异性分析。下载该物种所有种和亚种的序列文件,这里可以使用NCBI提供的ncbi_genome_download工具,以Streptococcus pneumoniae为例,下载该菌所有亚种有完整基因组的序列。ncbi-genome-download -s refseq -g "Streptococcus pneumoniae" bacteria -F fasta -l complete -p 16 -o Streptococcus_pneumoniae --flat-output#-s 可以选择refseq或者genbank#-g 下载的菌的名称#-F 表示下载的格式,一般选择fasta更贴合自己的需求#-l 表示下载基因组的类型,基因组有4种类型,complete', 'chromosome', 'scaffold', 'contig,之间的区别前面写过,也可以选择all这个参数。#-p 线程#-o 输出文件夹#--flat-output 表示不重新创建文件夹,下载的所有细菌基因组在一个文件夹中
下载完成后,查看目录下的该菌的个数。统计结果总计227个基因组序列。ll Streptococcus_pneumoniae/ |wc -l227
合并所有序列文件,形成一个fasta文件。该文件就是该菌的数据库序列文件,后续可以使用cd-hit,去掉重复相似度高的文件(该步骤的主要目的是缩小数据库的大小,提高运行效率,可选步骤),人工检查,去掉错误的序列(该步骤必须进行,特别是临床级别的应用要求,必须保证获得的序列是真实可靠的,没有错误干扰)等。
gunzip *.gzcat *.fna |sed -e 's/ /_/g' > ../all.fasta
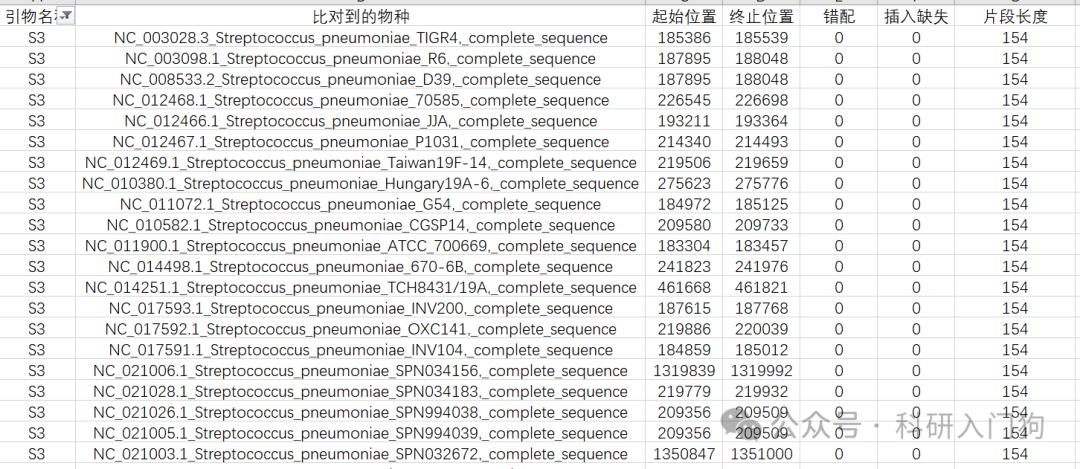
我利用这个数据库,就可以不用NCBI,初步的知道引物的覆盖度情况,我这里设计了3对Streptococcus pneumoniae的引物,利用blastn,将引物比对到该数据库。我设置的是容许3个错配,容许1个插入缺失、片段大小范围是50-1000。最后获得如下结果,可以了解该对引物比对到该菌的数据库情况。简单的统计了S1-S3,3对引物的覆盖度情况,3对引物都可以覆盖226个亚种,但是只有S3引物没有错配,完美匹配本数据库的索引肺炎链球菌基因组。
一个好的数据库,可以帮助我们做很多事情,提高工作效率。目前,搭建一个全和准的数据库,难度很大。但是搭建一个简易的数据库,也可以在初步的设计和分析中,给我们一个初步判定的结果。
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号