什么是第二代测序? 1953年,沃森和克里克在罗莎琳-富兰克林的DNA晶体学和X射线衍射基础工作的基础上确定了DNA的结构[1,2]。然而,第一个被测序的分子实际上是RNA--tRNA--在1965年由Robert Holley和后来的噬菌体MS2的RNA[3,4]。随后,各个研究小组开始将这些方法用于DNA测序,1977年Fredrick Sanger及其同事取得了突破性进展,开发了链式终止法[5]。到1986年,第一个自动化的DNA测序方法已经被开发出来[6,7]。这是测序平台发展和完善的黄金时代的开始,包括关键的毛细管DNA测序仪。 链式终止法,也被称为Sanger测序法,使用感兴趣的DNA序列作为PCR的模板,在延伸步骤中向DNA链添加修饰的核苷酸,称为双脱氧核苷酸(ddNTPs)[8]。当DNA聚合酶加入一个ddNTP时,延伸停止,导至产生无数的DNA序列的拷贝,所有长度都跨越了扩增片段。然后,在早期方法中使用凝胶电泳对这些链端寡核苷酸进行大小分离,或在后来的自动毛细管测序仪中使用毛细管进行分离,并确定DNA序列。随着这些巨大的技术进步,人类基因组计划于2003年完成[9]。2005年,第一个商业化的NGS平台,或已经成为第二代(2G)的平台问世,与桑格测序相比,能够以大规模并行的方式扩增特定DNA片段的数百万份[10]。 Sanger测序和2G NGS的关键原理有一些相似之处[11,12]。在2G NGS中,遗传物质(DNA或RNA)被分割开来,通过一个被称为适配序列连接的步骤,已知序列的寡核苷酸被连接在上面,使这些片段能够与选择的测序系统相互作用。然后,每个片段的碱基通过其发射的信号被识别出来。Sanger测序和2G NGS之间的主要区别来自于测序量,NGS允许并行处理数百万个反应,从而实现高通量、更高的灵敏度、速度和降低成本。大量的基因组测序项目,用Sanger测序方法需要很多年,现在用NGS可以在几小时内完成。 NGS技术有两种主要的方法,即短读和长读测序,每种方法都有自己的优势和局限性(表1)[13]。投资发展NGS的主要范围是它在临床和研究环境中的广泛适用性。在临床上,NGS通过鉴定种系或体细胞突变,被用来诊断各种疾病[14,15]。在临床实践中向NGS转变的理由是该技术的力量和持续下降的成本。NGS也是元基因组研究中的一个有价值的工具,用于传染病的诊断、监测和管理[16,17]。2020年,NGS方法在确定SARS-CoV-2基因组的特征方面发挥了关键作用,并在监测COVID-19大流行方面不断作出贡献[18,19]。
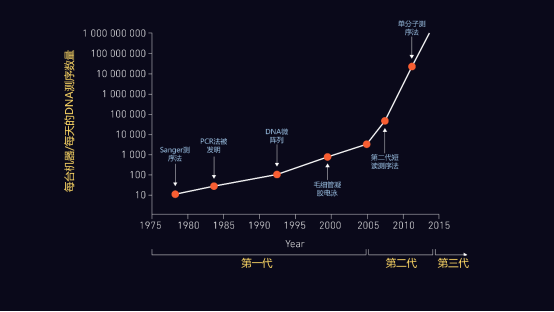
图1 | 测序方法学的演变 时间轴(X轴)表示第一代、第二代和第三代测序技术的引进,对照每台机器每天可测序的DNA千碱基数量(Y轴)。
第二代测序方法 NGS一词通常被认为是指2G技术,然而,第三(3G)代和第四(4G)代技术后来也发展起来,它们的工作原理不同。 2.1、测序平台/测序技术 第二代测序方法是成熟的,有许多共同的特点。然而,它们可以根据其基本的检测化学原理进行细分,包括连接测序(结合纳米球)和合成测序(SBS),后者进一步分为质子检测、火法测序和可逆终止器(图2)。
图2 | 代表2G测序平台的原理和不同化学方法的工作原理的图。 质子检测测序依赖于计算DNA聚合过程中释放的氢离子。与其他技术不同,它不使用荧光,也不使用改性核苷酸或光学。相反,pH值的变化是由半导体传感器芯片检测的,并转换成数字信息[20]。 火法测序利用检测焦磷酸的生成和光的释放来确定特定的碱基是否已被纳入DNA链中[21,22]。 到目前为止,最流行的SBS方法是利用‘桥式扩增’的可逆终止测序。在合成反应中,片段与流动池上的寡核苷酸结合,从序列的一侧(流动池上的P5寡核苷酸)到另一侧(P7)形成一个桥梁,然后进行扩增。添加的荧光标记的核苷酸用直接成像法检测[23]。 与SBS不同,连接测序不使用DNA聚合酶来创造第二条链。而是利用DNA连接酶对碱基配对错配的敏感性,用产生的荧光来确定目标序列。每次反应后拍摄的数字图像随后被用于分析。DNA纳米球测序是一种利用滚动循环复制的连接测序形式。合并的DNA副本被压缩成DNA纳米球,并以密集的斑点网格结合到测序玻片上,准备进行基于连接的测序反应[24,25]。虽然纳米球技术降低了运行成本,但产生的短序列对读图来说可能是个问题。 2G NGS技术总的来说比其他测序技术有一些优势,包括能够以快速、灵敏和经济的方式产生测序读数。然而,也有一些缺点,包括同族聚合物的解释能力差,聚合酶加入不正确的dNTPs,导至测序错误。短读长度也造成需要更深的测序覆盖率,以实现准确的等位基因组和最终基因组组装[26-30]。所有2G NGS技术的主要缺点是在测序前需要进行PCR扩增。这与文库制备(序列GC-含量、片段长度和虚假多样性)和分析(碱基错误/偏爱某些序列而不是其他序列)过程中的PCR偏倚有关。 3G测序的引入规避了对PCR的需求,对单分子进行测序,无需事先扩增步骤。第一个单分子测序(SMS)技术是由Stephen Quake及其同事开发的[31]。在这里,通过监测荧光标记的核苷酸与DNA链的结合,以单碱基的分辨率获得序列信息,并使用DNA聚合酶。根据不同的方法和使用的仪器,3G NGS的一些优势包括: ➤ 实时监测核苷酸的结合情况 ➤ 无偏倚测序 ➤ 更长的读取长度 然而,高成本、高错误率、大量的测序数据和低读深度都会带来问题[32,33]。 在4G系统中,3G的单分子测序与纳米孔技术相结合。与3G类似,纳米孔技术不需要扩增,采用的是单分子测序的概念,但集成了纳米级直径的微小生物孔(纳米孔),单分子通过这些孔并被识别。4G系统目前提供最快的全基因组序列扫描,但仍然相当昂贵,与2G技术相比容易出错,而且相对较新。因此,目前该技术可用的数据较少[34]。 2.2、2G测序方法和第 二代测序库准备的主要步骤 无论选择哪种2G NGS方法,有几个主要步骤必须根据目标(RNA或DNA)和选择的测序系统进行定制和优化。 2.2.1、样品制备(预处理) 从选定的样品(血液、痰液、骨髓等)中提取核酸(DNA或RNA)。对提取的样品进行质量控制(QC)检查,使用标准方法(分光光度法、荧光法或凝胶电泳法)。如果使用RNA,必须将其反转录为cDNA,然而一些文库制备试剂盒可能包括这一步骤。 2.2.2、文库制备 通常通过酶处理或超声处理,对cDNA或DNA进行随机片段化。最佳的片段长度取决于正在使用的平台。在优化这一过程时,可能需要在电泳凝胶上运行少量的片段样本。然后这些片段被末端修复并连接到更小的通用DNA片段上,称为适配体。适配序列有确定的长度和已知的寡聚体序列,与应用的测序平台兼容,在进行多重测序时可以识别。多重测序,每个样品使用单独的适配序列序列,使得大量的文库可以在一次运行中被汇集并同时进行测序。这种带有适配序列的DNA片段池被称为测序库。 然后可以通过凝胶电泳或使用磁珠进行大小选择,以去除任何太短或太长的片段,使其在选定的测序平台和协议上达到最佳性能。然后用PCR实现文库的富集/扩增。在涉及乳化PCR的技术中,每个片段都与一个乳化珠结合,这将构成测序簇的基础。扩增之后通常会有一个‘清理’步骤(例如,使用磁珠),以去除不需要的片段并提高测序效率。 最后的文库可以用qPCR进行质量控制检查,以确认DNA的质量和数量。这也将允许为测序准备正确浓度的样品。 2.2.3、测序 根据所选择的平台和化学方法,文库片段的克隆扩增可能发生在测序仪加载之前(乳剂PCR)或测序仪本身(桥式PCR)上。然后根据所选择的平台对序列进行检测和报告[35]。 2.2.4、数据分析 根据所使用的工作流程,对生成的数据文件进行分析。分析方法高度依赖于研究的目的[36-38]。 虽然它们可能会减少在一次运行中可以分析的样本量,但成对测序和配对测序在下游数据分析中提供了优势,特别是对于从头组装。这些技术将从一个片段的两端读取的测序读数连接在一起(成对的),或者被一个间隔的DNA区域分开(成对的)。 在选择测序策略时,显然有许多选择。以下是决定适当的文库制备和测序平台时的一些关键考虑因素: a) 所提出的研究问题 b) 样品类型 c) 短读或长读测序 d) DNA或RNA测序--你需要查看基因组还是转录组? e) 是需要全基因组还是只需要特定区域? f) 需要的读取深度(覆盖率)--针对具体的实验 g) 提取方法 h) 样品浓度 i) 单端、成对或配偶对读数 j) 需要的特定读数长度 k) 样品是否可以被复用? 生物信息学工具,取决于实验。根据样品和生物学问题,可以对整个序列分析过程进行调整。
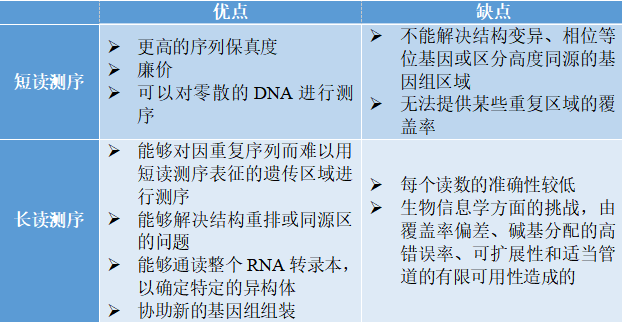
短读测序与长读测序 表1总结了短读测序和长读测序的优势和劣势。 表1 | 短读测序与长读测序的优势和劣势表
全外显子组与全基因组测序之间的区别是什么? 全基因组测序(WGS)是应用最广泛的NGS形式,指的是对基因组的整个核苷酸序列的分析。另一方面,全外显子组测序(WES)是一种有针对性的测序形式,只处理蛋白质编码的外显子。在人类中,这只占基因组的2%左右,因此提供了在这些区域进行更深入研究的机会。由于减少了测序负担,WES也可以提供比WGS更有成本效益的选择,并减少由此产生的测序数据的数量和复杂性。然而,由于只对基因组的一部分进行测序,重要的信息可能会被遗漏,新发现的机会也会减少。尽管成本增加,尽管迅速下降,以及相关的数据分析挑战,WGS因此提供了一个更强大的分析,可以揭示一个更完整的画面。
第二代测序数据分析 任何一种NGS技术都会产生大量的输出数据。序列分析的基本原理遵循一个集中的工作流程,其中包括原始读数质量控制步骤、预处理和绘图,然后是配准后处理、变体注释、变体调用和可视化。 对原始测序数据的评估对于确定其质量和为所有下游分析铺平道路是必不可少的。它可以对读数的数量和长度、任何污染性序列或任何低覆盖率的读数提供一个总体看法。计算测序读数的质量控制统计的最成熟的应用之一是FastQC。然而,对于进一步的预处理,如读数过滤和修剪,需要额外的工具。修剪读数末端的碱基和去除残留的适配序列序列通常能提高数据质量。最近,超快速工具已经问世,如fastp,它可以对测序数据进行质量控制、读数过滤和碱基校正,结合了传统应用的大部分功能,同时运行速度也比任何一个单独的工具快两到五倍[39]。 在对读数的质量进行检查并进行预处理后,下一步将取决于是否存在参考基因组。在新的基因组组装的情况下,产生的序列将使用它们的重叠区域排列成等位点。这通常是在处理管道的协助下完成的,这些管道可以包括脚手架步骤,以帮助等位基因组排序、定向和去除重复区域,从而提高组装的连续性[40,41]。如果将生成的序列与参考基因组或转录组进行映射(对齐),就可以确定与参考序列相比的变化。今天,有大量的绘图工具(超过60种),它们已经被调整为处理由NGS产生的越来越多的数据,利用技术进步和解决协议的发展[42]。由于映射器的数量不断增加,一个困难是能够找到最合适的映射器。信息通常分散在出版物、源代码(如果有的话)、手册和其他文件中。一些工具也会提供绘图质量检查,这是必要的,因为一些偏差只有在绘图步骤之后才会显示出来。与制图前的质量控制类似,正确处理制图读数是一个关键步骤,在此期间,重复的制图读数(包括但不限于PCR神器)被去除。这是一个标准化的方法,大多数工具有共同的特点。一旦读数被映射和处理,就需要以特定的实验方式对其进行分析,即所谓的变异分析。这一步可以识别单核苷酸多态性(SNPs)、缩减(碱基的插入或缺失)、倒位、单倍型、RNA-seq情况下的差异基因转录等等。尽管有许多用于基因组组装、比对和分析的工具,但仍需要不断推出新的和改进的版本,以确保其灵敏度、准确性和分辨率能够与快速发展的NGS技术相匹配。 最后一步是可视化,对其来说,数据的复杂性可能构成重大挑战。根据实验和提出的研究问题,有许多工具可以使用。如果有参考基因组,集成基因组查看器(IGV)是一个流行的选择[43],基因组浏览器也是如此。如果实验包括WGS或WES,变异体资源管理器是一个特别好的工具,因为它可以用来筛选数以千计的变异体,并允许用户专注于他们最重要的发现。像VISTA这样的可视化工具可以在不同的基因组序列之间进行比较。适合于从头开始的基因组组装的程序[44]比较有限。然而,像Bandage和Icarus这样的工具已经被用来探索和分析组装的基因组。
第二代测序的瓶颈 NGS使我们能够以以前不可能的方式发现和研究基因组。然而,NGS样品处理的复杂性暴露了管理、分析和存储数据集的瓶颈。主要挑战之一是测序数据的组装、注释和分析所需的计算资源[45]。NGS分析产生的海量数据是另一个关键挑战。数据中心的存储容量达到了很高的水平,并不断试图应对日益增长的需求,有可能出现永久性的数据丢失[46]。人们不断提出更多的策略,目的是提高效率,减少测序错误,最大限度地提高可重复性,确保正确的数据管理。
第二代测序的应用 自21世纪初以来,NGS已经成为现代医学研究和临床/诊断环境中以及药物发现方面的一个宝贵工具,包括WGS、WES、靶向测序、转录组、表观基因组和元基因组测序等方法的使用急剧增加。图3总结了针对不同数据集的工作流程和选项。
图3 | 表明不同样品类型的可能测序策略的流程图。 流程图表明不同样品类型,基因组、表观基因组和转录组,可能的NGS测序策略以及可能的工作流程。 通过WGS,研究人员不仅能够研究人类和动物的基因及其对疾病的参与,还能研究微生物和农业种群的特征,提供重要的流行病学和进化数据[47-52]。到目前为止,已经有大量的研究使用WGS鉴定突变、重排和融合事件。目前,WGS被用于抗菌素耐药性的监测,这是全球健康的主要挑战之一[53,54]。随着成本的不断降低,WGS被更多地用于临床样本的全人类基因组重测序,并可能很快成为临床实践中的常规[55]。最终,将需要WGS来为剩余的大部分基因组分配功能,并解读其在疾病中的作用。 它们更有针对性的性质使WES和定向测序成为人群和临床研究的有吸引力的选择[56,57]。尽管顾名思义有更多的局限性,但WES是个性化医学领域的一个重要临床工具。某些疾病的基因诊断,如癌症,以及其他疾病的基因特征,可以用这种方法以比WGS更经济的方式实现。 除了NGS在DNA测序方面的许多应用外,它还可以用于RNA分析。例如,这使得RNA病毒的基因组得以确定,如SARS和流感。重要的是,RNA-seq经常被用于定量研究,不仅有利于识别DNA基因组中的转录基因,还能根据RNA转录物的相对丰度识别它们的转录水平(转录水平)。DNA序列的潜在重排也可以通过识别新的转录物来确定[58,59]。 表观基因组测序可以研究由组蛋白修饰和DNA甲基化引起的变化。有不同的方法用于研究表观遗传机制,包括全基因组硫酸氢盐测序(WGBS)、染色质免疫沉淀(ChIP-seq)和甲基化依赖性免疫沉淀(MeDIP-seq),然后再测序[60,61]。根据所选择的方法,完整的DNA甲基组和组蛋白修饰图谱可以被绘制和研究,获得对基因组调控机制的洞察力。 元基因组测序可以为在特定环境中收集的样本提供信息。它能够比较混合微生物种群之间的差异和相互作用,以及宿主的反应。元基因组测序的一些潜在应用包括但不限于传染病诊断和感染监测、抗菌素抗性监测、微生物组研究和病原体发现[62]。 样品制备、测序技术和数据分析方面的技术进步意味着NGS也被用于单细胞水平,研究DNA、RNA和表观基因组的异质性和罕见变化。
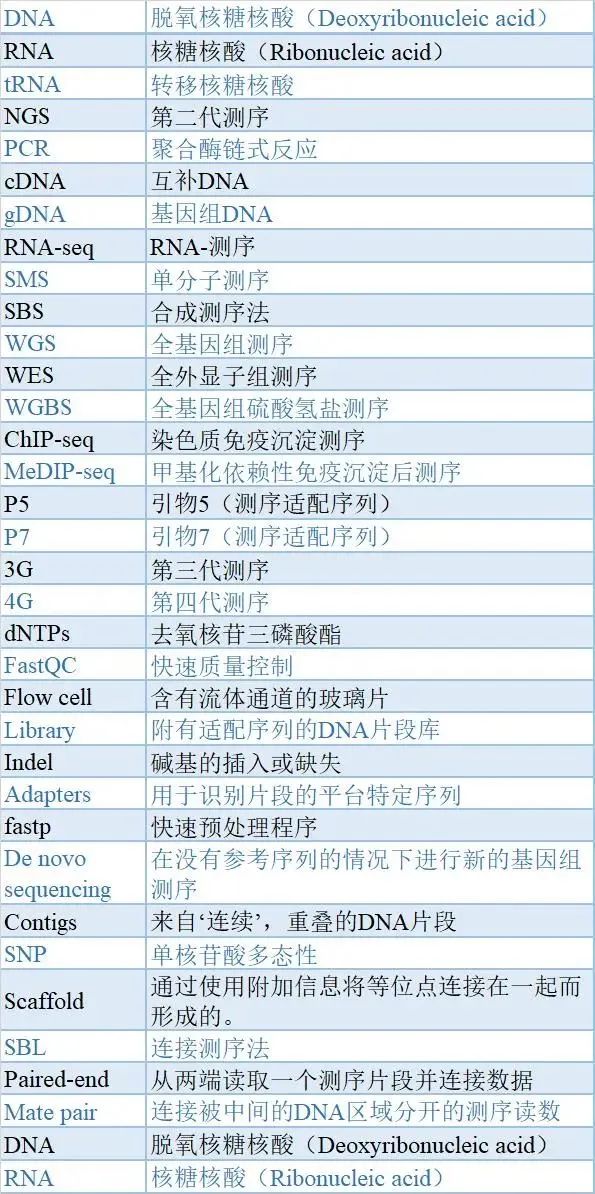
第二代测序的关键术语和缩略语 表2 | 与NGS有关的关键术语和缩略语。
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号