大家好,在此篇中和大家分享一下目前已上市FDA肿瘤大panel产品的体细胞突变检测算法。 一、前言 2022年9月初,国家卫生健康委临床检验中心(NCCL)发布了2022年《全国实体肿瘤高通量测序(大 Panel)肿瘤突变负荷检测室间质量评价预研结果报告》。本次共计收到62家实验室结果,基于评分方法,合格实验室共计49家。 肿瘤突变负荷(tumor mutational burden,TMB)一般指特定基因组区域内每兆碱基对(Mb)体细胞非同义突变的个数,因此检测TMB的准确性是直接和检测体细胞突变SNV的准确性相关。本次参评实验室使用的体细胞小突变检测工具如下图所示,主要分为使用自建工具或者是第三方软件。
二、FDA产品检测体细胞突变检测工具 FDA的产品的检测方法也分为使用自建方法和第三方软件,例如FoundationOne CDx使用自建的检测工具,MSK-IMPACT使用第三方软件。以下主要针对这2家的算法进行讲解。
FoundationOne CDx 为Foundation Medicine, Inc.的在2017年获得 FDA批准(FDA-approved,三类)的肿瘤大panel,以下简称F1CDx,F1CDx共包含324基因,全长2.2Mb,CDS区约0.8Mb。 2.1.1检测范围 碱基替换(substitutions),插入和缺失突变(InDels),拷贝数缺失(CNAs),融合突变(SV),微卫星不稳定(MSI)和TMB。 2.1.2检测工具 2.1.2.1碱基替换 碱基替换检测是使用贝叶斯方法进行的,该方法允许检测低突变等位基因频率 (MAF) 的新发体细胞突变,并通过结合组织特异性先验预期(prior expectation)提高对热点突变位点的检测灵敏度。将mapping quality <25或 base calls with quality ≤2的reads过滤掉。最终判断体细胞突变发生的标准为MAF ≥ 5%(热点为MAF ≥ 1%)。 具体算法公式如下所示: P(Mutation present| Read data “R”) = P(Frequency of mutation “F” > 0|R) = 1 – P(F = 0|R)
P(F = 0) = 1 – prior expectation “p” of the mutation in cancer type
P(R|F = i/n)的值使用每个候选突变位点观察到的等位基因count的多项分布去评估,使用观察到的error rates的经验值。如果P(“F” >0|R) > 99%时会作为候选的突变位点,最终当MAF ≥ 5% (MAF ≥ 1% at hotspots)时才报告为体细胞突变。在此之前,还需要使用Fisher’s test(P < 1e-6)去过滤strand bias,使用Kolmogorov–Smirnov test(P < 1e-6)去过滤read location bias ,此外如果在正常参考样本组中有2个或2个以上的正常样本中出现此位点突变的话,也会进行过滤。 2.1.2.2插入和缺失突变 为了检测插入缺失,使用 de-Bruijn 方法在每个目标外显子中进行从头局部组装(de novo local assembly)。涉及到的8个关键步骤如下所示: 1)收集至少一条read比对到目标区域的所有read-pairs。 2)将每条read分解成k-mers的小片段,并重新组装,由于变异位点的存在,组装最终会得到一个带有多个枝节点组装图,通过遍历整个组装图的所有可以通行的路径,可以得到多条长序列,这些长序列称之为haplotypes. 3)通过无间隙比对(ungapped alignment),将所有reads与每个候选haplotypes进行比较,并将每个read的read vote分配给具有最佳匹配的候选haplotype。候选haplotypes之间的关系通过拆分read vote来解决,由已经支持每个haplotype的reads数量加权。重复此过程,直到选择“获胜”haplotype。 4)将候选haplotype与参考基因组进行比对以报告突变位点。 候选插入缺失的过滤与碱基替换类似,根据经验针对repeats区提高等位基因频率的阈值,此外针对repeats相邻区域的质量指标如同GATK 中实施的相邻序列质量指标:相邻碱基不匹配百分比 < 25%,平均相邻碱基质量 > 25,read mismatches的平均数量≤ 2。最终判断标准为MAF ≥ 5%(热点 MAF ≥ 3%)。
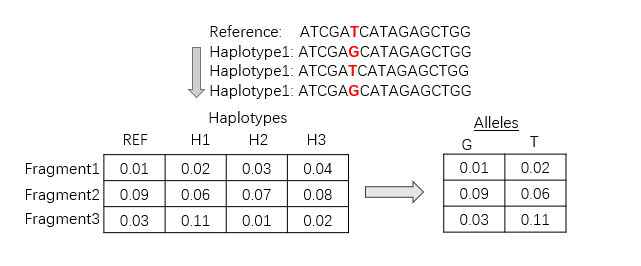
MSK-IMPACT 为Memorial Sloan-Kettering Cancer Center的获得 FDA批准的一款肿瘤大panel。 2.2.1 检测范围 碱基替换(substitutions),插入和缺失突变(InDels),微卫星不稳定(MSI)。 2.2.2检测工具 2.2.2.1 碱基替换 使用软件MuTect2 (version 1.1.4) 。 Mutect2算法如下所示: Mutect2在进行变异位点检测时的工作流程分为四大模块:Finding Active Regions(活跃区域鉴定),Assembling Haplotypes (基因序列局部重组),Somatic Genotyping和Filtering。 1) Finding Active Regions 活跃区域鉴定:遍历基因组数据,确定可能有变异位点的大致区域,称之为活跃区域(Active Regions),其他区域则是非活跃区域。如果没有感兴趣区间,就需要遍历全基因组进行活跃区间的鉴定,如果有感兴趣区间,则只需要在感兴趣区间内进行活跃区域鉴定。 2) Assembling Haplotypes 局部组装,通过建立de Bruijn-like graph 对于活跃区域下的所有reads进行局部重新组装,由于变异位点的存在,组装最终会得到一个带有多个枝节点组装图(graph)。同时根据reads序列的碱基质量信息以及每个位点变异的频率,还需要对图的各种枝节点进行部分修剪。通过遍历整个组装图的所有可以通行的路径,可以得到多条长序列,这些长序列称之为haplotypes. 3) Somatic Genotyping 对于每个活跃区域,统计该区域内的所有reads以及对区域内reads重新组装得到的haplotypes,利用Pair-HMM算法将每条read和每条haplotype分别对齐,计算两两序列之间匹配的相似度。由此可以得到一个相似度矩阵,矩阵记录每条read对应上每条Haplotype的匹配相似度(read-haplotype likelihoods)。 Mutect2将此矩阵再转换成fragment-haplotype 矩阵,通过对paired reads增加log-likelihoods即P(fragment|haplotype) = P(read 1|haplotype)P(read 2|haplotype),fragment即指的是一对paired-end reads之间的片段。对于每一个候选变异位点,将fragment-haplotype矩阵再转换成fragment-allele的矩阵,即选出覆盖该位点的fragment和haplotypes对应的相似度子矩阵如下图左侧所示。将每个Frament的allele的似然值设置为包含该allele的haplotype的似然值中的最大似然值。对上图右侧的每条fragment的allele的相似度值矩阵值,应用贝叶斯法则来计算该位点的每种基因型的概率,其中概率最大的就是该位点的基因型。
4) Filtering 根据以上三个步骤找到候选变异位点之后,还需要对变异位点进行筛选。变异位点筛选是为了过滤掉测序错误,数据污染等原因造成的假阳性变异,以及过滤掉生殖细胞变异。过滤器(FilterMutectCalls)基于Mutect2计算出来的变异位点的各种相关数值信息是否大于阈值对变异位点进行过滤。 2.2.2.2 插入和缺失突变 使用软件SomaticIndelDetector (一款GATK的工具包 version 2.3.9) |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号