富含α-Syn的聚集物的存在是共核蛋白病的标志,这是神经退行性疾病的一个子集,包括帕金森(PD)、路易体痴呆(DLB)和多系统萎缩(MSA)。遗传学研究已经证实位于染色体4q21–23上编码α-Syn的SNCA基因的突变和倍增,导至早发家族性帕金森(PD),并在大脑中广泛沉积α-Syn聚集体。α-Syn在病理过程中,发生构象变化,形成富含β-sheet的结构,包括寡聚体、原纤维和不溶性纤维,最终积聚在路易小体(LBs)中。尽管传统上定义疾病的是蛋白质聚集的不溶性终末期物种,但具有显著毒性的是可溶性中间低聚物,可诱导异常的钙信号,产生活性氧、线粒体功能障碍和神经细胞死亡。α-Syn低聚物形成的早期动力学过程由于其本质上的瞬态、异质性和低丰度而难以研究。单分子荧光方法可用来观察蛋白质的相互作用和构象,这些方法广泛依赖于检测足够接近时两个荧光团之间的激发能量转移。利用smFRET,研究团队先前在体外跟踪了α-Syn聚集,区分聚集过程中形成的低聚物的不同结构群,并比较了SNCA突变的低聚体形成动力学。 近日,来自英国的一组研究团队在杂志Nature Neuroscience上发表了一篇题为“Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity”的文章,在本研究中,研究团队整合了敏感的生物物理方法——FRET biosensor、smFRET和三维(3D)FRET-CLEM,以精确跟踪α-Syn聚集动力学,并可视化神经元内的早期聚集反应。在这里,研究团队展示了脂质膜上的早期事件如何触发聚集,并揭示了突变α-Syn在线粒体中聚集并改变线粒体功能的机制,最终导至毒性。
图片来源:Nature Neuroscience 主要内容 FRET biosensor测定α-Syn聚集 为了对α-Syn进行可视化,在氨基酸残基90位点(A90C)突变处引入荧光标记,该突变位于结构的外围,对α-Syn的聚集和毒性的影响可以忽略不计,一个标记Alexa Fluor 488(α-Syn-AF488),另一个标记了Alexa Fluor 594(α-Syn-AF594)。594 nm直接激发信号可以观察到总α-Syn在细胞内的积累,无论α-Syn的聚集状态如何。488 nm激发供体荧光团后,通过检测受体荧光团(AF549)的信号,可以观察到低聚物的形成(FRET信号)。 用500 nM α-Syn(含约1%低聚物和约99%单体)处理原代神经元:α-Syn-的摄取导至直接信号(总α-Syn)和FRET信号(低聚物形成),二者都随着时间的推移强度增加,反映了α-Syn-的持续摄取和细胞内随后的聚集(图b)。 只使用α-Syn单体(没有低聚物)的情况下,在初始滞后阶段后,FRET信号强度增加(图c)。3 小时以内,500 nM单体诱导FRET信号(低聚物形成),而50 nM 在48小时诱发FRET信号。WT和所有突变体的总α-Syn摄取量和FRET信号增加呈现时间依赖性(图d)。A53T在直接激发信号和细胞内FRET信号方面表现出最大的增加,这表明与其他突变相比,摄取和低聚化增加。
FRET biosensor检测A53Tα-Syn的快速细胞内聚集。图片来源:Nature Neuroscience 以FRET效率评估结构变化 smFRET可以确定体外结构从毒性较小、松散相关的“A型”低聚物转变为毒性大、抗蛋白酶K、富含βsheet的“B型”低聚物。利用此处检测到的细胞内FRET信号,研究团队通过计算细胞内聚集物的FRET效率(E)来确定低聚物的不同FRET群体是否可以在神经元内形成。 WTα-Syn的FRET效率随着时间的推移而增加,表明该蛋白可以在细胞内寡聚和结构转化。在早期时间点(3 小时和3 天),仅FRET效率低的低聚物群体存在。3天之后 ,第二批FRET效率较高的低聚物开始出现。从单体到低聚物的组装发生得很快(<3 小时),而转化为高FRET低聚物的时间长达数天。A53T、A30P和E46K突变体也显示了细胞中类似的结构转换。
FRET效率可评估结构变化。 图片来源:Nature Neuroscience A53T显示出加速聚集 A53T细胞(1%低聚物和99%单体)表现出快速组装,A53T单体处理的细胞在0–30分钟 也表现出快速组装成小的低聚物,并在24小时中持续增大。 细胞内FRET、smFRET和TIRF显微镜可以检测细胞内自组装、低聚物形成和转化为蛋白酶K抗性B型低聚物的初始阶段。细胞内的聚集是浓度和时间依赖性的,细胞内的滞后期减少。与WT和其他突变体相比,A53T的聚集速率增加,部分原因是A53T摄取增加和细胞内浓度增加。
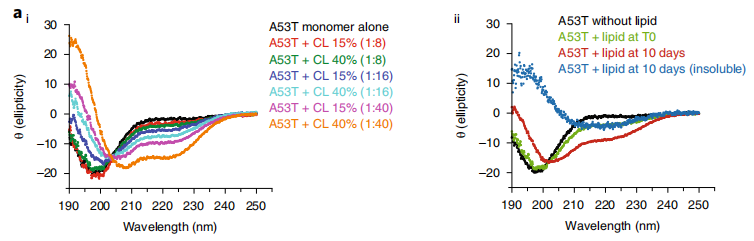
A53T显示出加速聚集。 图片来源:Nature Neuroscience 心磷脂加速A53T的寡聚化 研究团队研究了α-Syn和含有心磷脂(CL)的100 nm大小脂质小泡之间的相互作用。结果表明,α-Syn在含有CL的囊泡存在的情况下首先采用α-螺旋构象。随着CL含量和脂:蛋白摩尔比的增加,检测到α-螺旋结构增加。随着时间的推移,α-螺旋结构逐渐转变成一系列的蛋白质结构混合物,其中β-sheet含量显著。这些数据证实,α-Syn最初与CL相互作用并形成二级结构(α-螺旋),但随着时间的推移,逐渐转变为β-sheet。另有数据显示,A53Tα-Syn与脂质小泡的相互作用不仅导至CL的提取,还导至CL并入淀粉样纤维,并且这些聚集物与CL的共定位增加。 综上所述,这些数据表明,CL可以快速触发A53T的聚集,然后CL被纳入α-Syn的聚集结构,这可能会进一步加速聚集过程。
心磷脂加速A53T的寡聚化。 图片来源:Nature Neuroscience A53T诱导线粒体功能障碍 研究团队用复合物I底物NADH的自体荧光测定细胞氧化还原状态和复合物I功能。发现500 nM A53T诱导NADH自发荧光增加(119 ± 2.48%),表明复合物I功能被抑制。A53T诱导的复合物I抑制与线粒体去极化相关(120.4 ± 2.6%)。A53T处理的细胞与WT处理的细胞相比,ATP生成显著减少,且增加了超氧化物的生成速率。
A53T诱导线粒体功能障碍。 图片来源:Nature Neuroscience A53Tα-Syn诱导mPTP开放, mROS加速寡聚化和细胞死亡 线粒体通透性转换孔(mPTP)的早期开放介导了α-Syn低聚物诱导的细胞毒性。下一步研究团队测试了A53Tα-Syn是否影响mPTP开放。结果显示A53Tα-Syn降低了mPTP打开的阈值(图a)。mPTP的开放需要α-Syn转变成富含β-sheet寡聚体的结构构象。因此,A53T单体与线粒体膜接触,迅速形成寡聚体,并打开mPTP,诱导细胞凋亡和细胞毒性。 A53T α-Syn生成mROS,研究团队还研究了mROS是否改变聚集。结果显示mROS的生成是A53T α-Syn细胞内寡聚化的关键因素,清除mROS可以调节聚集物的形成。 这些结果表明,呼吸受损和mROS生成与CL协同作用,驱动A53T单体的细胞内聚集并导至神经元死亡。
A53Tα-Syn诱导mPTP开放,mROS加速聚集和细胞死亡。图片来源:Nature Neuroscience SNCA-A53T hiPSC衍生神经元的细胞表型 从两名携带A53T突变(SNCA-A53T)的患者、一名iso-CTRL患者和两名健康志愿者(CTRL)的hiPSCs中生成皮层神经元。与对照神经元相比,SNCA-A53T iPSC衍生的神经元积累并分泌更多具有渗透能力的内源性寡聚物。与对照细胞相比,SNCA-A53T神经元显示出较高的细胞内FRET信号和总α-Syn摄取量(图c)。并且SNCA-A53T神经元表现出异常的线粒体功能,类似于直接应用A53Tα-Syn后观察到的线粒体功能:膜电势差降低(图d),mROS产生增多(图e),复合I抑制,PTP早期开放。
SNCA-A53T hiPSC衍生的神经元表现出线粒体功能障碍。图片来源:Nature Neuroscience 总结与讨论 在这项研究中,研究团队使用单分子Förster共振能量转移(smFRET)生物传感器追踪了α-Syn的细胞内构象状态,结果表明,α-Syn-在神经元中以浓度依赖和序列特异的方式从单体状态转化为两种不同的寡聚状态。线粒体脂质心磷脂触发A53Tα-Syn的快速聚集,心磷脂被装配在聚集的脂蛋白复合物中。线粒体聚集物损害复合物I的活性并增加线粒体活性氧(ROS)的生成,从而加速A53Tα-Syn的低聚化并导至线粒体膜的通透性和细胞死亡。这些过程也在PD患者诱导的多能干细胞(iPSC)衍生的含有A53T突变的神经元中观察到。研究团队的结果强调了线粒体膜新生α-Syn聚集的机制和随后的神经毒性。 在此,研究团队揭示了线粒体促进α-Syn聚集的两个关键机制。(1)A53Tα-Syn在细胞内外结合CL;(2) CL是A53Tα-Syn快速聚集形成纤维的有效触发因素;以及(3)在CL诱导的聚集过程中,CL在形成时被包含在蛋白质聚集物中,这种脂蛋白共组装可以作为进一步表面诱导伸长的反应物。除了为聚集提供脂质种子外,线粒体也是活性氧的重要来源。A53Tα-Syn抑制复杂的I依赖性呼吸,损害ATP生成并使线粒体膜去极化,导至ROS生成增加。氧化环境反过来又促进了神经元中A53T单体的进一步聚集。总之,这些作用使线粒体易于PTP的早期开放和caspase 3依赖性凋亡的激活。 总之,这个研究阐明了α-Syn早期聚集过程机制,并有望为临床上治疗PD提供新的思路和理论基础。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号