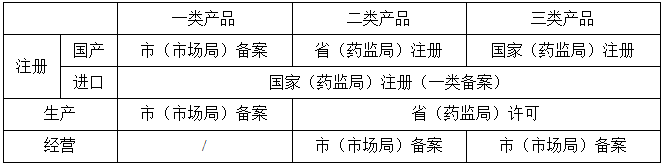
这是刘博谈合规的第十一篇,在本篇中,我们来介绍新版医疗器械监督管理条例亮点之二,风险管理理念贯穿医疗器械全生命周期。 02 风险管理理念贯穿医疗器械全生命周期 对于医疗器械监督管理的基本原则之一,就是风险管理,这在新版条例的第五条中进行了规定。 News 第五条 医疗器械监督管理遵循风险管理、全程管控、科学监管、社会共治的原则。 对于风险管理,首先在分类上就进行了控制,我国目前的一二三类产品,就是按照低中高风险来进行分类的,具体见第六条。 News 第六条 国家对医疗器械按照风险程度实行分类管理。 第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。 第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。 第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。 评价医疗器械风险程度,应当考虑医疗器械的预期目的、结构特征、使用方法等因素。 国务院药品监督管理部门负责制定医疗器械的分类规则和分类目录,并根据医疗器械生产、经营、使用情况,及时对医疗器械的风险变化进行分析、评价,对分类规则和分类目录进行调整。制定、调整分类规则和分类目录,应当充分听取医疗器械注册人、备案人、生产经营企业以及使用单位、行业组织的意见,并参考国际医疗器械分类实践。医疗器械分类规则和分类目录应当向社会公布。 对于一类、二类和三类产品,注册、生产、经营的管理层级是不一样的,而进口和国产产品的注册管理层级又是不一样的,具体可见下表。
表1|不同分类产品的管理层级 新版条例在这方面也做了很多的规定,比如注册方面的规定包括条例的第十三条、第十五条和第十六条; News 第十三条 第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。 医疗器械注册人、备案人应当加强医疗器械全生命周期质量管理,对研制、生产、经营、使用全过程中医疗器械的安全性、有效性依法承担责任。 News 第十五条 第一类医疗器械产品备案,由备案人向所在地设区的市级人民政府负责药品监督管理的部门提交备案资料。 向我国境内出口第一类医疗器械的境外备案人,由其指定的我国境内企业法人向国务院药品监督管理部门提交备案资料和备案人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。未在境外上市的创新医疗器械,可以不提交备案人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。 备案人向负责药品监督管理的部门提交符合本条例规定的备案资料后即完成备案。负责药品监督管理的部门应当自收到备案资料之日起5个工作日内,通过国务院药品监督管理部门在线政务服务平台向社会公布备案有关信息。 备案资料载明的事项发生变化的,应当向原备案部门变更备案。 News 第十六条 申请第二类医疗器械产品注册,注册申请人应当向所在地省、自治区、直辖市人民政府药品监督管理部门提交注册申请资料。申请第三类医疗器械产品注册,注册申请人应当向国务院药品监督管理部门提交注册申请资料。 向我国境内出口第二类、第三类医疗器械的境外注册申请 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号