近年来,随着分子诊断技术的升级迭代,分子诊断的临床应用越来越广泛和深入,分子诊断市场进入快速发展期。 笔者总结市场上常见的分子诊断技术,分为上中下三篇,上篇介绍PCR技术,中篇介绍核酸等温扩增技术,下篇介绍测序技术。 PCR技术 PCR (polymerase chain reaction)聚合酶链式反应,是体外DNA扩增技术之一,至今已经超过30年的历史。 PCR技术于1983年由美国Cetus公司的KaryMullis首创,1985 年Mullis申请了PCR专利,同年在Science上发表了第一篇PCR 学术论文,1993 年Mullis因此获得了诺贝尔化学奖。 PCR基本原理 PCR可以将目标DNA片段扩增一百万倍以上,其原理是在DNA聚合酶催化下,以母链DNA为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板DNA互补的子链DNA的过程。
标准 PCR 过程分为三步: 1.变性(Denaturation):利用高温使DNA 双链分离。DNA双链之间的氢键在高温下(93- 98℃)被打断。 2.退火(Annealing):在 DNA 双链分离后,降低温度使得引物可以结合于单链DNA 上。 3.延伸(Extension):DNA 聚合酶由降温时结合上的引物处开始沿着DNA 链合成互补链,延伸完成,则完成一轮循环,DNA片段数增加一倍。 往复循环这三个步骤25-35 次,DNA 片段数将得到指数级增加。
PCR的巧妙之处在于针对不同的目标基因可以设计不同的引物,使目标基因片段在短时间内得到百万级的放大。 目前为止,PCR可以分为三类,分别是普通PCR、荧光定量PCR和数字PCR。 第一代普通PCR 采用普通PCR 扩增仪来对靶基因进行扩增,然后采用琼脂糖凝胶电泳对产物进行检测,只能做定性分析。 第一代PCR主要缺点:
第二代荧光定量PCR 荧光定量PCR(Real-Time PCR),也叫做qPCR,通过在反应体系中加入能够指示反映进程的荧光探针,通过荧光信号的积累来监测扩增产物的积累,通过荧光曲线来判断结果,并可以借助Cq 值和标准曲线来定量。
qPCR 技术由于操作过程在封闭体系中进行,降低了污染概率,并且可以通过对荧光信号监测从而进行定量检测,因此临床应用最为广泛,已成为PCR中的主导技术。 实时荧光定量PCR所使用的荧光物质可分为:TaqMan荧光探针、分子信标和荧光染料。
1)TaqMan荧光探针: PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。 探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。
2)SYBR荧光染料: 在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料非特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。SYBR仅与双链DNA进行结合,因此可以通过溶解曲线,确定PCR反应是否特异。
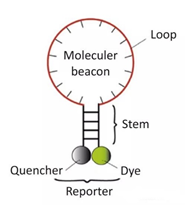
3)分子信标 是一种在5和3末端自身形成一个8个碱基左右的发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对,导至荧光基团与淬灭基团紧紧靠近,不会产生荧光。
PCR产物生成后,退火过程中,分子信标中间部分与特定DNA序列配对,荧光基因与淬灭基因分离产生荧光。
第二代PCR主要缺点:
第三代数字PCR 数字PCR(DigitalPCR, dPCR, Dig-PCR)通过终点检测计算目标序列的拷贝数,无需采用内参和标准曲线即可进行精确的绝对定量检测。 数字PCR采用终点检测,不依赖于Ct值(循环阈值),所以数字PCR反应受扩增效率的影响降低,对PCR反应抑制物的耐受能力提高,具有很高的准确度和重现性。 由于具备高灵敏度、高精确度的特点,不易被PCR反应抑制剂干扰,无需标准品可实现真正意义的绝对定量,而成为研究和应用热点。 根据反应单元的不同形式,主要可分为微流体式、芯片式和微滴式三大类系统。 1)微流体数字PCR,Microfluidic digital PCR,mdPCR。 基于微流控技术,对DNA模板进行分液,微流控技术能实现样品纳升级或更小液滴的生成,但液滴需要特殊吸附方式再与PCR反应体系结合,mdPCR已逐渐被其他方式取代。 2)微滴数字PCR,Droplet-based digital PCR,ddPCR。 利用油包水微滴生成技术对样品进行微滴化处理,将含有核酸分子的反应体系分成成千上万个纳升级的微滴,其中每个微滴或不含待检核酸靶分子,或者含有一个至数个待检核酸靶分子。 以伯乐的ddPCR为例,主要包括三个步骤:
第一,准备样本和生成微滴,如下图,8x3的微孔板,一排加样本,一排加油滴,通过微滴生成器每个样本生成20000个微滴。
第二,进行“油包水”PCR,把微孔板放入PCR扩增仪,对每个微滴进行40个热循环的PCR反应。
第三,读取微滴结果,把微孔板放入读取仪,通过流式细胞技术获取每个微滴PCR终点结果的荧光信号,并用泊松分布原理纠正结果。
Bio-rad去年上市的Bio-Rad QX ONE数字PCR系统整合了微滴生成、热循环反应及微滴读取等步骤,最大程度减少人工操作。 配备四个独立的荧光检测通道,结合ddPCR特有的高阶多重PCR技术,可以在单反应孔中实现8重拷贝数变异(CNV)检测、5重突变检测或4重基因表达检测。
3)芯片数字PCR,Chip-baseddigital PCR,cdPCR。 利用集成流体通路技术在硅片或石英玻璃上刻上许多微管和微腔体,通过不同的控制阀门控制溶液在其中的流动,将样本液体分成大小一致的纳升级于反应孔种进行数字PCR反应,实现绝对定量。 以法国Stilla technologies 公司的cdPCR技术为例,主要包括以下步骤:
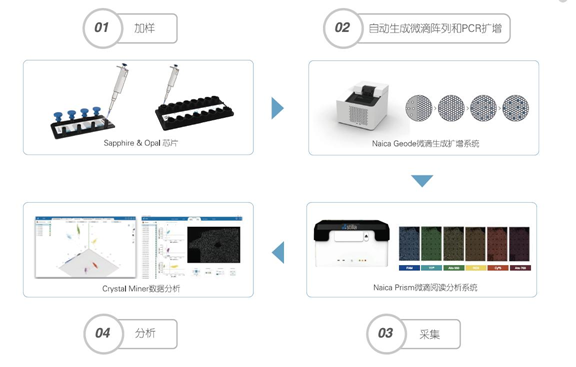
第一,加样和生成微滴:将样本和PCR反应液加入微流控芯片,放入仪器中,仪器可通过物理方法生成单层平铺的20000~30000个微滴阵列。 第二,对每个微滴进行PCR扩增:在Naica Geode微滴生成扩增系统自动进行PCR扩增。 第三,读取和分析结果:将芯片置于Prism微滴读取分析系统上进行荧光信号采集,计数阴阳性微滴,通过泊松分布计算获得靶标基因绝对拷贝数浓度。
总结一下,将样本和PCR反应液加入微流控芯片,放入仪器中,通过物理方法生成单层平铺的微滴阵列,随后对每个微滴进行扩增,然后进行六通道荧光信号采集,计数阴阳性微滴,通过泊松分布计算获得靶标基因绝对拷贝数浓度。 第三代PCR主要缺点:
PCR的各种延伸技术 1.递减PCR(touchdown PCR):前几循环温度逐渐下降。 2.逆转录PCR(RT-PCR):以由mRNA逆转录而来的cDNA 为模板,也因为是从表现型基因来进行增量的,由此产生出来的cDNA 产物不带有内含子(基因中不具意义的段落),常应用于分子克隆技术。 3.热启动PCR(hotstart PCR):以高热激活型核酸聚合酶进行反应,减少非专一性产物。 4.巢式PCR(nested PCR):先用低特异性引物扩增几个循环以增加模板数量,再用高特异性引物扩增。 5.多重PCR(multiplex PCR):在同一个管中使用多组引物。 6.复原条件PCR(reconditioning PCR):PCR 产物稀释 10 倍后重新放入原浓度的引物和dNTP 等循环 3 次,以消除产物中的异二聚体。 7.dsRNA 合成(dsRNA replicator):合并使用high-fidelity DNA polymersae、T7RNA聚合酶与Phi6 RNA replicase;从双股 DNA转录为对应的双股RNA(dsRNA)。可应用于RNAi实验操作。 8.COLD-PCR (co-amplification at lower denaturation temperature PCR):用以检测突变或特殊等位基因的PCR 应用技术。 参考文献: 1.刘婉彤等,分子诊断技术的临床应用进展 2.杨柳等,非鳞非小细胞肺癌基因检测技术研究进展 3.G.Terrance Walker,etc.Strand displacement amplification-an isothermal, in vitro DNA amplification technique 3.各公司官网 以上图片来源于各公司官网及公开渠道,如有侵权请联系我们删除。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号