体外诊断试剂产品注册技术审评报告
产品中文名称:胚胎植入前染色体非整倍体检测试剂盒(半导体测序法) 产品管理类别:三类 6840 申请人名称:苏州贝康医疗器械有限公司
国家食品药品监督管理总局 医疗器械技术审评中心
基本信息
苏州工业园星湖街 218 号生物纳米园 A3 楼 101 单元申请人生产地址:苏州工业园星湖街 218 号生物纳米园 B3 楼 201 单元;A3 楼 101 单元受托生产企业生产地址:广州高新技术产业开发区荔枝山路 6 号,1号楼 4 楼 401-408 室;2 号楼 1 楼、4 楼
产品审评摘要
表 1 试剂盒主要组成成分
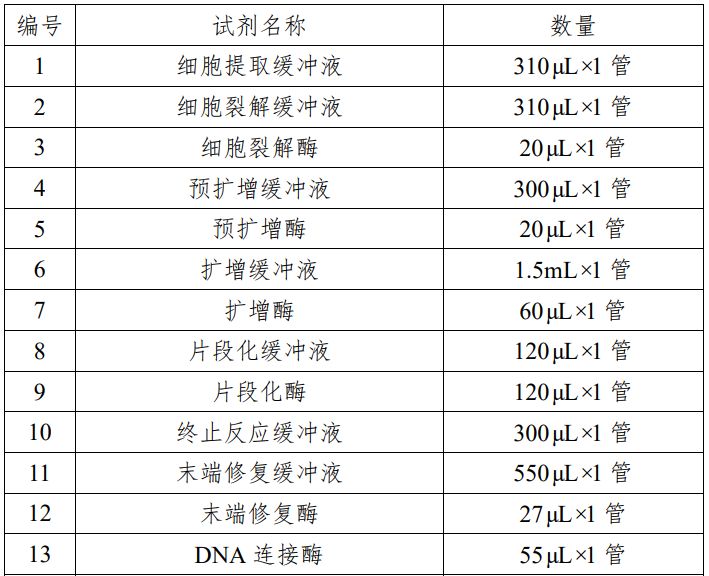
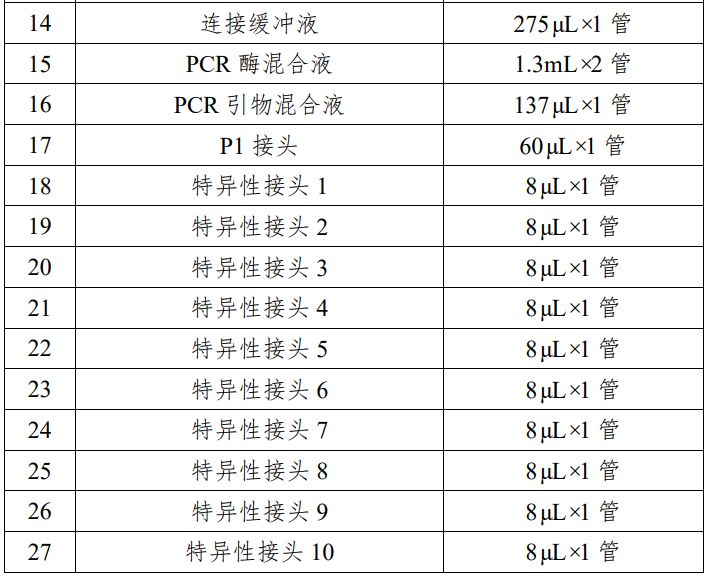
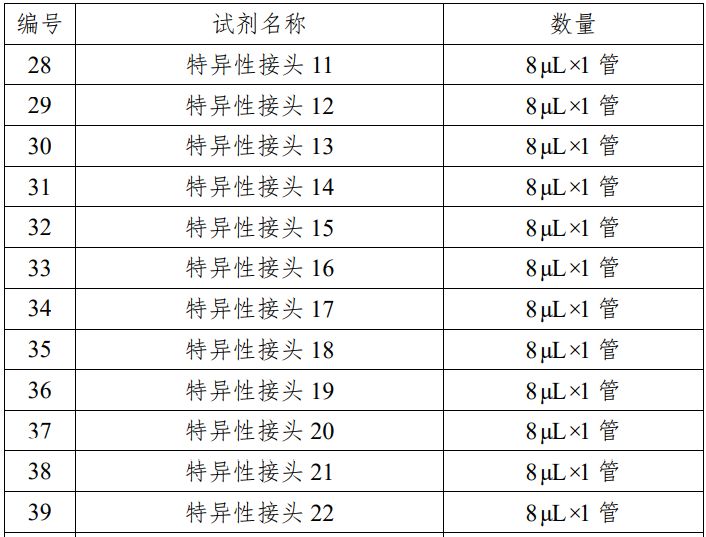
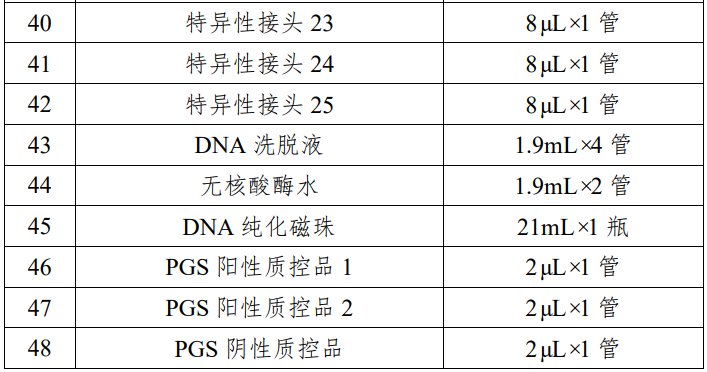
本产品用于定性检测试管婴儿过程中体外培养胚胎的囊胚滋养层细胞的脱氧核糖核酸(DNA),通过对胚胎部分细胞的 DNA 进行检测,分析胚胎染色体是否存在非整倍体数量异常,辅助临床医生判断胚胎是否植入,本产品用途为构建测序文库。 本产品适用于女性年龄 35 岁及以上进行试管婴儿的患者;夫妻双方或一方存在染色体异常的患者;三次以上的试管婴儿植入失败者;三次以上自然流产患者;生育过染色体异常患儿的夫妇。 检测结果仅供参考,不单独作为确诊的依据,本产品不用于拷贝数变异的检测。 该试剂盒是利用单细胞全基因组扩增技术对从囊胚期胚胎获得的细胞进行扩增,利用半导体高通量测序技术对扩增产物进行全基因组测序,通过生物信息软件对测序结果进行数据分析,判断胚胎是否存在染色体非整倍体异常。 对囊胚期胚胎进行活检,采用优化的简并寡核苷酸引物 PCR 技术对活检细胞进行全基因组扩增,将扩增产物随机打断为 200bp 左右的 DNA 片段,在 DNA 片段两端加上测序接头,构建测序文库。通过基于半导体高通量测序技术的 DA8600 基因测序仪进行全基因组测序,利用生物信息学软件对测序结果进行分析,得到匹配到每条染色体上的有效序列数量,计算有效序列数量与参考数据库中相应染色体序列数量的比值,若该比值过高,则该染色体可判断为三体或重复;若该比值过低,则该染色体可判断为单体或缺失,实现对染色体非整倍体异常的检测。该试剂盒主要原材料包括:细胞裂解酶、预扩增酶、扩增酶、片段化酶、末端修复酶、DNA 连接酶、PCR 酶混合液、PCR 引物混合液、P1 接头及特异性接头 1-25 和质控品等,这些原材料均为外购方式获得,质控品由细胞系供应商提供,并由申请人分离细胞后制备获得。申请人对主要原材料进行了供应商选择,通过功能性试验,筛选出最佳的原材料供应商,制定了主要原材料质量要求并经检验合格。 企业参考品包括阳性参考品、阴性参考品、嵌合体参考品、数据量控制参考品。阳性参考品 18 份,是由多种染色体非整倍体阳性细胞样本组成;阴性参考品 5 份,是由染色体非整倍体阴性细胞样本组成;嵌合体参考品 6 份,是由两种不同染色体拷贝数变异样本混合制备的细胞样本组成;数据量控制参考品 1 份,由 YH 细胞样本组成。 质控品是由 2 份阳性质控品和 1 份阴性质控品组成,用于检测过程中试剂和仪器的质量控制。其中阳性质控品是由染色体非整倍体阳性细胞样本(21 三体和 XO)组成;阴性质控品是由染色体非整倍体阴性细胞样本组成。 申请人通过使用初步确定的配方进行反应体系配制,对反应体系中细胞裂解酶、预扩增酶、扩增酶、片段化酶、末端修复酶、连接酶、PCR 酶等进行筛选和优化,通过功能性试验和生产工艺研究,确定了最佳的生产工艺和反应体系。 分析性能评估内容包括染色体非整倍体阳性符合率、微缺失微重复符合率、阴性符合率、嵌合体符合率、数据量控制、重复性、不同实验室间精密度、高通量测序配套试剂验证、分析特异性。 1.染色体非整倍体阳性检测符合率:对连续三批生产的试剂盒采用 13 份染色体非整倍体阳性样本进行检测,检测结果表明染色体非整倍体阳性符合率为 100%;使用国家参考品中的非整倍体阳性参考品检测,符合率 100%。 2.微缺失微重复检测符合率:该产品对微缺失微重复的检出情况进行了分析性能评估;使用国家参考品中的微缺失微重复参考品检测,符合该指标。3.阴性检测符合率:对连续三批生产的试剂盒采用 10 份阴性样本进行检测,检测结果表明阴性符合率为 100%;使用国家参考品中的阴性参考品检测,符合率 100%。4.嵌合体检测符合率:对连续三批生产的试剂盒采用 6 份嵌合样本进行检测,检测结果表明对 30%嵌合的嵌合体样本符合率达到30%以上;70%嵌合体符合率达到 60%以上;使用国家参考品中的嵌合体参考品检测,符合该指标。5.数据量控制检测:对连续三批生产的试剂盒采用 1 份企业数据量控制参考品进行检测,检测结果表明数据量控制参考品的检测有效数据量不低于 1M,基因组覆盖率不低于 4%;使用国家参考品中的数据量控制参考品检测,符合该指标。6.重复性:对连续三批生产的试剂盒分别进行三次上述的染色体非整倍体阳性检测符合率实验、微缺失微重复检测符合率实验、阴性检测符合率实验、嵌合体检测符合率实验、数据量控制检测实验,检测结果表明重复性均满足技术要求规定的性能指标。7.不同实验室间精密度:对连续三批生产的试剂盒采用企业参考品分别在两个不同的实验室进行检测,每批试剂盒检测三次,检测结果表明不同实验室间重复性检测均满足技术要求规定的性能指标。8.高通量测序配套试剂的验证:使用广州市达瑞生物技术股份有限公司的测序反应通用试剂盒(半导体法)(备案号粤穗械备20160061),对企业参考品进行三次重复检测,检测结果表明该测序反应通用试剂盒可以作为该试剂盒的配套测序试剂。9.分析特异性:对连续三批生产的试剂盒采用 57 份染色体微缺失微重复样本进行检测,均未检测出非整倍体,检测结果表明分析特异性达到 100%。申请人采用已知染色体非整倍体阴性样本构建参考数据库,然后采用已知的染色体非整倍体阴性样本和染色体非整倍体阳性样本进行参考区间的确定,最终确定的染色体非整倍体阴性样本的参考区间为[-0.51,0.37],染色体非整倍体阳性样本的参考区间大于 0.37、小于-0.51。具体过程如下,通过 240 例染色体非整倍体阴性样本的检测结果构建参考数据库,使用该试剂盒对 234 例染色体非整倍体阴性样本和染色体非整倍体阳性样本进行检测,分析这些样本每条染色体上检测到的序列数目,并计算该序列数目与参考数据中对应染色体的序列数目的比值,即 CNV 值,通过对不同 CNV 值(拷贝数变异 CopyNumber Variation, CNV)区间的划分,统计分析真阳性率、假阳性率、真阴性率以及假阴性率,从而最终确定该试剂盒的参考区间。 申请人对该试剂盒的加速稳定性、长期稳定性、冻融稳定性、开瓶稳定性、运输稳定性进行了研究,确定了在各种条件下该试剂盒的有效保存时间。另外对临床样本进行了稳定性、冻融次数研究,确定了临床样本的有效保存时间。加速稳定性实验:取连续三批生产的试剂盒,将除片段化酶、末端修复酶、DNA 连接酶及质控品以外的试剂解冻后置于 37℃恒温培养箱中,分别存放 1 天、2 天、3 天、4 天后取出,对试剂盒的性能指标进行检测,确定该试剂盒在不超过 37℃条件下可稳定存放 3 天。 长期稳定性实验:取连续三批生产的试剂盒,将试剂盒置于储存环境中,分别于第 0、2、4、6、8 个月取出,对试剂盒的性能指标进行检测,确定该试剂盒在其储存环境下的有效期为 6 个月。冻融稳定性实验:取连续三批生产的试剂盒,将试剂盒由储存环境中取出,在室温条件下充分溶解后继续置于储存环境下。如此反复冻融 4、6、8、10、12、14 次,对试剂盒的性能指标进行检测,确定该试剂盒可以反复冻融 12 次。 开瓶稳定性实验:取连续三批生产的试剂盒,将试剂盒开瓶后置于储存环境中,于 2、4、5、6 个月对试剂盒的性能指标进行检测,确定该试剂盒开瓶后可以稳定存放 5 个月。运输稳定性实验:取连续三批生产的试剂盒,将试剂盒按照储存温度要求采用冷链运输,运输时长为 5 天,分别于运输结束后及有效期末对试剂盒的性能指标进行检测,确定该试剂盒运输时限为 5 天。 样本稳定性的研究:申请人对胚胎细胞样本的稳定性、冻融次数进行了研究,确定样本的长期保存条件为-80℃冰箱,保存时间不超过 18 个月,冻融次数不超过 5 次。 申请人在山东大学附属生殖医院、中信湘雅生殖与遗传专科医院、中国医科大学附属盛京医院、第四军医大学唐都医院、兰州大学第一医院、南京市妇幼保健院共六家临床试验机构开展了临床试验。临床试验采用考核试剂与金标准进行比较研究的试验方法,验证考核试剂的临床有效性。染色体非整倍体阳性金标准对比验证方法为荧光原位杂交(FISH);染色体非整倍体阴性金标准对比验证方法为染色体核型分析,包括产前诊断羊水穿刺核型分析、流产组织核型分析或新生儿脐带血核型分析。每例纳入统计的样本均采用考核试剂和金标准对比验证方法进行检测。 考核试剂临床试验共入组 1482 对符合适应症的受试者,检测6282 例胚胎样本,检测出染色体非整倍体阳性胚胎样本 1672 例,染色体非整倍体阴性胚胎样本 4483 例。阳性样本进行了全部 23 对染色体非整倍体异常的 FISH 验证,共 381 例,FISH 验证结果与本试剂盒检测结果一致,其中,Chr13、Chr16、Chr18、Chr21 以及性染色体阳性验证例数为:Chr13(35 例)、Chr16(36 例)、Chr18(26 例)、Chr21(46 例)、性染色体(29 例);阴性胚胎样本有 1162 例进行了移植,750 例已经确定妊娠,获得 291 例核型分析检查结果(其中羊水穿刺核型分析 273 例,新生儿脐带血核型分析 14 例,流产组织核型分析 4 例),核型分析结果与待考评试剂盒检测结果一致。 通过对 381 例 FISH 验证确认的阳性样本以及 291 例核型分析验证确认的阴性样本统计分析,待考评试剂盒的灵敏性为 100%(95%CI:99.00%~100%),特异性为 100%(95%CI:98.70%~100%)。进行 Kappa 一致性检验,Kappa=1(P<0.001),显示已检测样本中,考核试剂与金标准对照方法具有良好的检测一致性。 利用基于二项分布的统计学算法对所有检测样本进行阳性预测值和阴性预测值统计分析,本临床试验的阳性预测值为100.00%(95%CI:98.88%~100.00%),阴性预测值为 100.00%(95%CI:98.46%~100.00%)。由于本临床试验无法对所有阴性样本以及阳性样本全部进行验证的特点,通过对受试者的基线特征和入组胚胎的基线特征进行分析,受试者基线和入组胚胎基线在已验证与未验证样本中均无具有临床意义的显著差异,可以认为在本产品检测结果的统计中,完成验证的样本能够代表总体样本。 临床可接受的准确性下限估计:根据 2002 年卫生部颁发的《胎儿染色体核型分析技术规范》要求核型分析的准确率不低于 98%,因此认为,如本类产品准确性达到 98%,则可以认为该产品达到了临床诊断的准确性要求。经计算,在准确度下限设置为 98%时,仍允许阳性和阴性样本中出现验证失败样本,但在已完成验证的样本中,未出现 1 例验证失败,即可以认为即使完成所有样本验证,本临床试验仍能满足准确性要求。 综上所述,该产品临床试验资料对产品的临床性能进行了全面研究,临床试验结果基本符合临床应用要求。申请人需在该产品上市后进一步完成以下工作:上市后需继续搜集至少 10 家临床机构、全部临床使用数据作为临床补充资料在产品下一次延续注册时提交,继续收集各染色体检测异常情况(本试剂检测阳性)及植入后(本试剂检测阴性)结果情况,植入后的胚胎继续追踪其与金标准检测结果(核型分析/出生随访)的对比情况。该项临床资料应当由出具数据各临床机构签章。根据“YY/T 0316-2016 医疗器械风险管理对医疗器械的应用”标准,对该产品进行风险分析。 本产品用于定性检测试管婴儿过程中体外培养胚胎的囊胚滋养层细胞的脱氧核糖核酸(DNA),通过对胚胎部分细胞的 DNA 进行检测,分析胚胎染色体是否存在非整倍体数量异常,辅助临床医生判断胚胎是否植入。本产品适用于女性年龄 35 岁及以上进行试管婴儿的患者;夫妻双方或一方存在染色体异常的患者;三次以上的试管婴儿植入失败者;三次以上自然流产患者;生育过染色体异常患儿的夫妇。上述人群是胚胎染色体非整倍体高发人群,通过检测结果来辅助判断胚胎是否植入,减少因植入异常胚胎而造成的反复种植失败、反复流产、出生缺陷等,减少患者生理、心理伤害及社会经济负担。 该试剂盒检测结果会受到样本来源、样本采集过程、样本运输条件等样本因素的影响,同时也受到实验操作、实验环境、试剂储存等试验因素的影响,可能导至数据质量降低或检测失败。使用者须了解检测过程中可能存在的潜在风险及检测的局限性,详见产品说明书中【检验方法的局限性】。 不符合该试剂盒说明书中【样本要求】的样本或不恰当的实验操作会导至数据质量降低或检测失败,请严格按照产品说明书中【样本要求】及【检验方法】的要求操作和进行实验过程质控。 该试剂盒在检测过程中涉及基因扩增,在非可控的实验室操作可能由于环境中气溶胶的存在导至结果不可靠,同时 PCR 操作过程中气溶胶的泄露可能会导至设备甚至实验室的污染。因此,请在可控的实验室进行检测操作,操作人员必须进行专业培训,严格按照说明书操作。 在现有技术条件下,该产品检测结果仅辅助医生判断胚胎是否植入,胚胎植入后还将按照体外辅助生殖相关诊疗规程对植入胚胎的情况进行产前检测和产前诊断等。 通过环境控制、生产监控、成品检验和增加说明书警示内容等防范措施,对该试剂盒的已知和可预见的安全风险进行控制和降低,剩余风险可以被控制在可接受范围内,同时没有带来新的危害与安全风险。在目前认知水平上,认为该试剂盒上市带来的受益大于风险。尽管目前认为该试剂盒的受益大于风险,但是为保证用械安全,基于对主要剩余风险的防控,已在该试剂盒说明书中提示以下信息: 1.预期用途:该试剂盒适用于女性年龄 35 岁及以上进行试管婴儿的患者;夫妻双方或一方存在染色体异常的患者;三次以上的试管婴儿植入失败者;三次以上自然流产患者;生育过染色体异常患儿的夫妇。2.警示及注意事项:该试剂盒说明书中明确了该试剂盒【检验方法的局限性】及使用中的【注意事项】。
综合评价意见 本申报项目为境内第三类医疗器械产品注册,属于创新审批项目(编号:2015168)。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第 680 号)、《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令 2014 年第 5 号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。申请人在该产品上市后需继续搜集至少 10 家临床机构、全部临床使用数据作为临床补充资料在产品下一次延续注册时提交,继续收集各染色体检测异常情况(本试剂检测阳性)及植入后(本试剂检测阴性)结果情况,植入后的胚胎继续追踪其与金标准检测结果(核型分析/出生随访)的对比情况。该项临床资料应当由出具数据各临床机构签章。 | 

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 














