
2018年,是医疗器械GMP全面铺开的元年,也是医疗监管加强加严的关键之年,尤其以长春长生生物的疫苗案为标志性事件,医疗制造行业的合规掀起了一场自顶层而下的风暴。在2019的新年伊始,医疗人咖啡就带您回顾一下2018年国家局的飞检情况,用一串串数据来辨清监管形势,透析法规要点。
1、2018年到底有多少家器械生产企业被国家局飞检?
纵观NMPA新网站的飞行检查一栏,飞检部队还是蛮拼的,2018年足迹遍布23个省,共88家器械生产企业受到检查。通过下图可以看出,NMPA偏爱江、浙、沪和北京、天津、山东,因为这些都是器械大省啊。
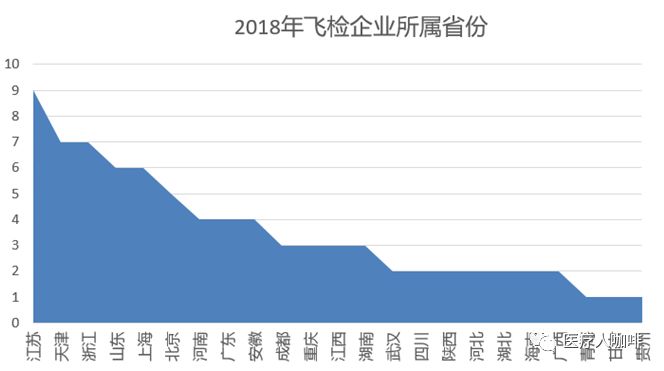
如果细究起来,88家企业当中,有20家企业正处于停产状态。也可以理解为,飞检就是要看看这些停产企业是否真的停产,还是故弄玄虚。值得一提的是,虽然陕西仁康药业有限公司处于停产状态,但是也被从查出了6条一般缺陷,看来停产企业只要记录还在,仍处于飞检监控之下。本文就主要围绕着有检查缺陷的69家企业说开来去。
2、一般企业被查都有多少缺陷?

3、有多少家停产整顿,又有多少限期整改?
69家企业,1家处于停产状态,18家责令停产整改,50家限期整改。

4、GMP条款,企业最容易犯什么错?
2018年的飞检缺陷共754条,分别落在《医疗器械生产质量管理规范》、“无菌附录”、“植入附录”、“体外诊断试剂附录”和“义齿附录”的156个条款上。

缺陷对应条款,预料之中主要是与规范相关的条款,涉及542条,其次是无菌105条。

考虑缺陷的集中度,对重复10次出现的条款号进行梳理,发现有20个条款最容易出现问题,总共出现缺陷404项,占总数的53.6%。 


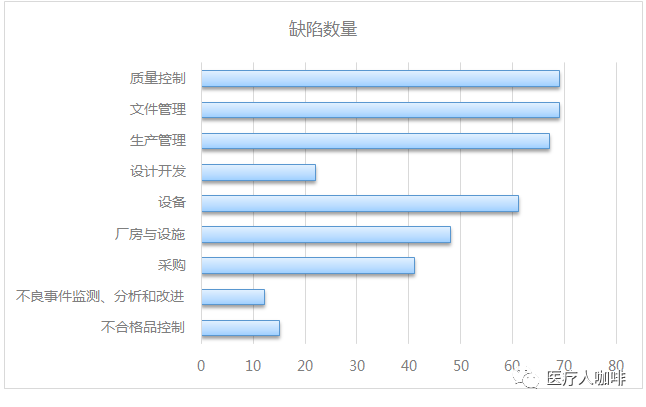
其中,质量控制、文件管理、生产管理和设备设施是重灾区。
5、停产整顿的企业要害问题是什么?

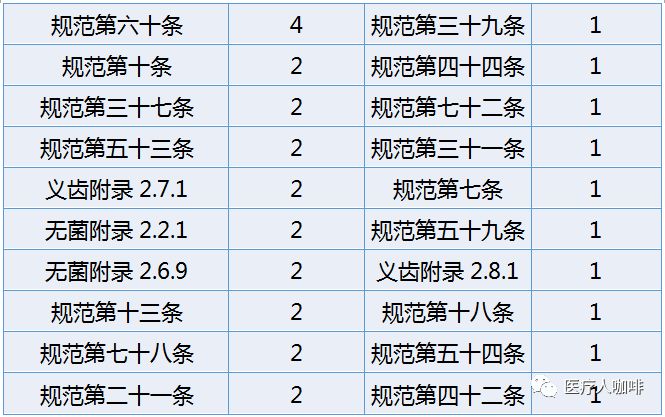
上表可以看出,规范第五十八条实为飞检的杀手锏。
“第五十八条 企业应当根据强制性标准以及经注册或者备案的产品技术要求制定产品的检验规程,并出具相应的检验报告或者证书。
需要常规控制的进货检验、过程检验和成品检验项目原则上不得进行委托检验。对于检验条件和设备要求较高,确需委托检验的项目,可委托具有资质的机构进行检验,以证明产品符合强制性标准和经注册或者备案的产品技术要求。”
到底这12项严重缺陷是哪些呢,详见下表。 序号 | 内容 | 1 | 产品检验规程未涵盖经注册的产品技术要求中的如下性能指标:弯曲性能、弯曲弹性模量、X射线阻射性能、拉伸粘接强度、剪切粘接强度、组成成分及百分含量。 | 2 | 未能严格按照国家强制性标准制定出厂检验规程及检验。1)高频漏电流测试,GB9706.4要求负载电阻200欧姆,测试电阻200欧姆,线间距50cm。检测人员操作用负载电阻400欧姆,测试电阻200欧姆,线间距25cm进行测试。2)生产过程中抽查电缆耐压试验,操作规程要求10s升到1000V,保持1min;实际是20s升到1000V,保持40s。在成品检验中,高频漏电流测试要求10s升到1000V,保持1min;实际在18s上升到4000V,保持42s。 | 3 | 企业制定的环氧乙烷残留量检验方法要求,检测时应分别配制1μg/ml-10μg/ml的6个浓度的标准曲线后进行检测,但检查发现:1.灭菌日期为2018年8月3日某批号产品的环氧乙烷残留量检验企业仅配制5个浓度标准曲线;
灭菌日期为2018年4月28日生产的某批号产品的环氧乙烷残留量检验,企业配制的标准曲线所用溶液浓度为1μg/ml-40μg/ml。 | 4 | 批号20170214的一次性使用无菌阴道扩张器检验报告书中“无菌(生物菌片)试验原始记录”显示,采用菌片试验方法检验无菌项目,与检验依据YZB/贛2327-2013《一次性使用无菌阴道扩张器》的无菌检验方法不一致。 | 5 | 医用钬(Ho:YAG)激光治疗机成品检验报告(报告编号006)中“纤芯直径、长度”、“光纤传输效率”、“光纤传输效率不稳定度”的测试结果为“符合”,但相应的检验原始记录中无实际检测数据。 | 6 | 抽查企业的生产批号为1805QGH29A01(灭菌批号为18E00429B-1)的产品检验报告,EO残留量、无菌的出厂检验所用样品为灭菌替代品,但《EO残留量检验操作规程》、《无菌检验操作规程》规定抽取12个产品组件替代品(EO残留量2个、无菌10个)进行检验,未明确具体组件替代品。 | 7 | 1.产品技术要求中规定直径、后顶焦度测量按GB/T11417规定的方法进行,国标中明确规定,直径测量时“将镜片放在测池内,维持测池内标准盐溶液的温度为20℃±0.5℃。转动镜片,3次独立测量最大和最小直径” ;后顶焦度测量时“在测量前,水凝胶镜片浸入20℃±0.5℃的标准盐溶液中平衡至少30min。”。现场核查企业测量时使用常温纯化水代替标准盐溶液,与企业产品技术要求规定不符;
2.企业提供的成品检验规程中性能指标有5.5无菌要求、5.7保存液渗透压,抽查出厂检测报告(生产批号20180720、20180719)中缺少无菌、保存液渗透压检测项目。抽查出厂检验报告(生产批号20170518)无菌检测项目判定为合格,而未提供相应的原始无菌检测记录。 | 8 | 二氧化碳激光治疗机注册产品标准规定产品瞄准光波长应为635nm±20nm,但企业未根据此要求制定相应检验规范,且未配备相应检测设备。 | 9 | 1.旧版的硅凝胶乳房植入体成品检验规程未包含壳体D4、D5,硅凝胶小分子残留物质、边缘夹角检验项目。2.企业2018年6月对检验规程进行了修订,修订后的检验规程未包含该产品技术要求中的2.4乳房植入体成品性能“边缘夹角”指标。 | 10 | 1.企业出具的一次性使用无菌阴道扩张器的出厂检验报告“中抗变形能力”项目均为“符合要求”,但未见该项目检验原始记录。
2.抽查2018年生产的扩张器批检验记录,批号71804002、71805002产品检验报告依据为“YY 0336-2002”(现行版本为YY 0336-2013);批号71803009、71806003产品检验报告依据为“产品技术要求”,产品技术要求中无菌的试验方法GB/T14233.2-2005,YY 0336-2013中无菌试验方法为《中国药典》。企业的试验方法不一致。 | 11 | 产品出厂检验报告(批号170329 编号170329-001)中的检验项目与产品出厂检验规程不一致,缺少检验规程中“工作情况检验”5.1的检验项目。 | 12 | 产品检测时部分项目未依据YY0300—2009《牙科学 修复用人工牙》标准进行:孔隙和其他缺陷项目,标准规定试样制备时应采用低速锯或湿研磨装置在冷却条件下截取平面,实际使用陶瓷砂轮干法磨削截取平面;色泽及融合性项目,未按标准规定布置反射率(30±5)%的漫射灰背景等测试条件进行检查 |
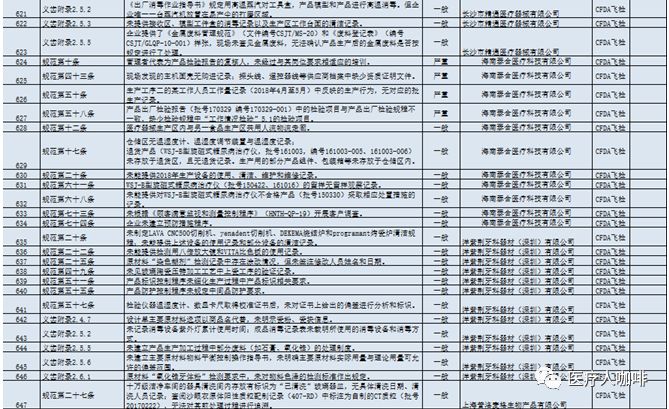
从上严重项可以看出,检验项目的缺失、检验方法与文件规定不一致、检验设备配置不到位、检验记录不完整是最容易一招毙命的。 为了使大家更好的了解飞检缺陷情况,韩老师通过几天的整理,编制了2018飞检缺陷汇总(EXCEL版),对缺陷进行了统计和标准化,方便大家进行学习借鉴。欢迎大家加入医咖俱乐部,通过俱乐部获取最新资料。 


| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 









