



一、概述
上市前批准(PMA:premarket approval)程序是针对Ⅲ类医疗器械评估其安全性和有效性的。Ⅲ类医疗器械是能够在支撑和维持人体生命健康方面起实质性作用,并且存在潜在和不明原因风险的医疗器械。由于Ⅲ类医疗器械本身存在的风险,FDA认为一般和特殊控制不足以保证其安全性和有效性,因此,这类产品需要申请PMA程序。
FDA会根据PMA申请所提交的数据能否科学有效的证明该器械符合预期用途,以此来决定该申请能否通过。FDA法规规定会在180天内完成PMA审核并做出能否上市的最终决定。但实际上,时间通常要长一些。在做出最终决定前,专家委员会会在一次公开会议上对申请进行审核,并给出该申请是否可以通过的意见。在FDA通知申请人最终决议后,通知和连同该决定所依据的数据会张贴在网站上,此后30天内,相关责任人若不满意结果,可请求重审。
二、申请PMA的条件
PMA适用于Ⅲ类医疗器械,Ⅲ类医疗器械可以通过在产品分类目录中查到。美国有专门供公众查询的产品分类目录数据库。下图就是FDA关于产品类别的查询界面。可以通过输入产品的名称和产品代码对产品进行查询。产品的数据库每周日会定期进行更新。FDA已经为1700多种医疗器械规定了类别,并根据评审专家将其分成了20种医疗大类,点开review panel就可以看到,分别是麻醉学、普通医疗、耳鼻喉、普通手术器械、免疫学、眼科、放射科、细血管、泌尿肠胃、微生物学、骨科、临床化学、神经科、病理学、毒物学、牙科、血液学、妇产科、物理医学、分子遗传学。
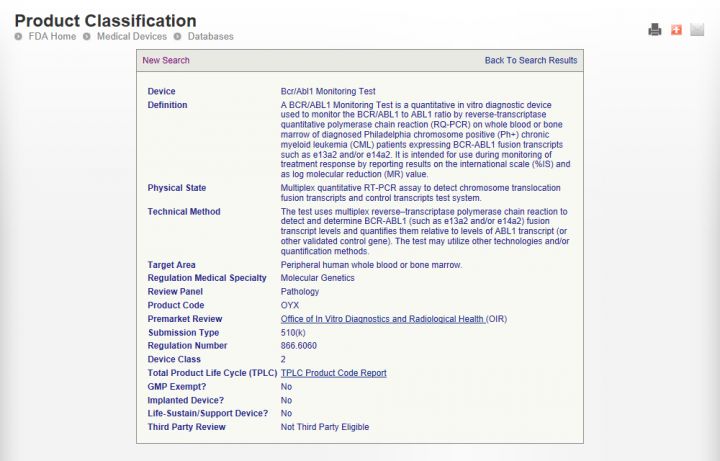
值得注意的是,申请PMA的器械会包括一些新器械,这种情况下,分类数据库中只标注了这类产品的类型名称和产品代码。产品代码是由三个大写的字母组成,例如Bcr/abl 基因检测产品代码是OYX。如下图所示。
因此如果不确定产品是否应该申请PMA,将产品代码输入到PMA数据库或510(K)数据库中进行搜查。确定510(K)数据库中并未出现与之实质性等同的产品。然后确定其是否应该走PMA申请程序。另外,新类型的产品,其不能在分类数据库中找到,而且其具有Ⅲ类医疗器械所具有的特征。那么其必须申请PMA。下图是PMA数据库。
三、PMA审查程序
1. PMA的审查程序包括了四个步骤:
1)行政审查所递交材料的完整性;
2)技术、法规、质量方面的实质性审查;
3)专家委员会的审查和建议;
4)FDA给出最终的审查结果。
第一步的行政审查,FDA办事员审核资料是否完整。确认资料完整后,才算资料正式递交,此后方可进行第二步的实质性审查。一般来讲,FDA会在收到PMA申请的45天内,邮件通知申请人资料是否通过行政审查。若FDA确定接收申请者的PMA申请(这个过程叫做filing a PMA),会告知申请人PMA编号(PMA reference number)和接收日期。申请人可以根据此PMA编号来查审查进度。国外的很多编号前都会加“reference”。例如客户编号是“customer reference number”;订单编号是“order reference number”,“reference”在这种情况下可以翻译为“供查阅的”,也可不翻译。接收日期就是PMA开始接受180天审查的日期。
除了PMA申请被接收,还有被FDA拒绝的情况,如果申请人提交的资料是以下几种情况,则很可能会被FDA驳回申请。
第一,申请资料不完整,不符合FD&C Act(联邦食品药品法案)515(c)(1) (A)-(G)节关于PMA所需递交资料的要求。那么PMA递交资料到底包括哪些呢?主要包括下表中所列出的13项内容。

第二,PMA没有完整包含Sec 814.20下要求的项目,并且没有充分说明资料缺少的理由。CFR 814.20是关于PMA申请的资料要求。
第三,申请人对于该设备有待定的510(k)程序,并且FDA尚未确定该设备是否属于Sec 814.1(C)的范围。CFR 814.1是关于PMA的应用范围要求。
第四,PMA申请中包含虚假的重要事实陈述。
第五,PMA未附有21 CFR 54所规定的财务证明。
在接受PMA申请后,FDA会进行第二步的实质性审查。在审查过程中,FDA会通过主要或者次要缺陷信函通知申请人,以获取FDA完成审查所需的任何信息。申请人可以在提交PMA后100天内要求与FDA会面,讨论该申请的审核状态。关于“第100天会议”程序可以在相关指导原则中找到。(Guidance on PMA Interactive Procedures for Day-100 Meetings and Subsequent Deficiencies - for Use by CDRH and Industry; Final)
如果申请人主动或根据FDA的要求提交PMA补正内容,该补正内容包含先前未报告的研究中的重要新数据和先前报告的研究的重要更新数据,或者对先前提交的数据的详细的新分析,或以前遗漏的重要信息,审查期可延长至180天。

 /3
/3 





 浙公网安备33010802005999号
浙公网安备33010802005999号


