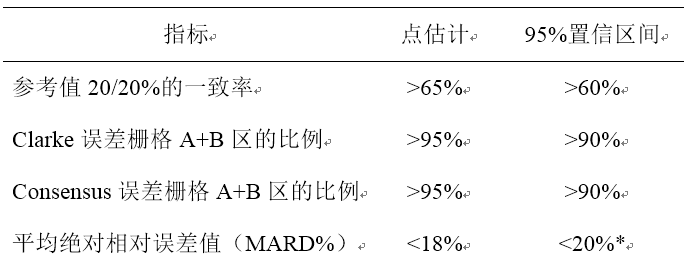
持续葡萄糖监测系统在原有成人适用范围的基础上新增申报儿童的适用范围,如何进行临床评价? 持续葡萄糖监测系统(Continuous Glucose Monitoring System,CGMS)目前批准适用范围一般为“产品用于糖尿病成年患者(≥18岁)的组织间液葡萄糖水平的连续或定期监测。产品可提供并存储连续葡萄糖值,供用户跟踪葡萄糖浓度变化的趋势,如葡萄糖水平低于或高于预设值,产品可发出提醒。产品测量结果不作为决定和调整糖尿病患者治疗方案的依据。”国际儿童和青少年糖尿病学会(ISPAD)发布的《2022 ISPAD临床实践指南:儿童、青少年和年轻糖尿病患者的血糖目标和血糖监测》中推荐将儿童、青少年及25岁以下年轻患者纳入持续葡萄糖监测(CGM)控制目标。已上市用于成人的持续葡萄糖检测系统拟增加2-17岁儿童适用范围时,在已有成人适用范围批准的基础上,需通过临床试验路径进行临床评价。 2-17岁儿童可分为2-5岁、6-17岁2个年龄组。其中,6-17岁年龄组入组样本量应保持与成人临床试验要求一致,以静脉血糖值(使用EKF血糖检测仪或Yellow Spring Instrument(YSI)的检测结果)为对照,至少应入组60例提供1200个测量点。2-5岁年龄组儿童如考虑伦理学因素可采用指尖血测量血糖值作为对照,应入组一定样本量的受试者以保证主要评价指标满足要求。 临床试验可采用多中心的自身对照设计,主要评价指标包括:1.与参考值的20/20%的一致性,通过一致率表示;2.测量点落在Clarke误差栅格分析A+B区的比例;3.测量点落在Consensus误差栅格分析A+B区的比例;4.平均绝对相对误差值(MARD%)。 2-5岁、6-17岁各年龄组临床试验结果要求上述四项主要评价指标均应同时达到表1所列标准,方能认为产品性能符合临床应用需要,即按照共同主要终点设置。上述四项主要评价指标中有任何一项或多项未达到所列出的评价标准要求,则不认可产品性能。需注意的是,MARD%评价需先计算每位受试者水平的结果,而不能像其他指标直接计算所有测量点水平的汇总结果。 表1主要评价指标对应的评价标准及原则
*评价95%置信区间的上限,其余指标评价下限。 上述关于持续葡萄糖监测系统临床试验设计的要求,仅针对传统持续葡萄糖检测系统,即产品用于糖尿病患者的组织间液葡萄糖水平的连续或定期监测,测量结果不作为决定和调整糖尿病患者治疗方案的依据。 病原体抗体定性检测试剂应如何理解产品检出限浓度? 根据《定性检测体外诊断试剂分析性能评估注册审查指导原则》,建议申请人采用对已知分析物浓度的样本进行系列稀释后重复检测的方法,确定申报试剂的检出限(一般为95%阳性检出率),并在该检测限浓度水平对常见分析物型别进行验证。 上文中“检出限”或“检测限浓度水平”,指经其他可靠方法或可给出量值的试剂(如定量试剂、可给出连续量值信号的其他定性试剂)确定的具体数值,以考察申报试剂的检出能力和可复现性。 进口体外诊断试剂提交变更注册时,如何提交原产国说明书? 如涉及到说明书变更,应提交变更前后的原文说明书,即原产国上市说明书。不应仅提交变更前后中文说明书的英文翻译件或公证版的中文说明书。 体外诊断试剂稳定性评价常见问题探讨 体外诊断试剂稳定性是指体外诊断医疗器械在制造商规定界限内保持其性能特性的能力,反映了产品能够随着时间的推移保持一致的性能特征。稳定性研究一般包括实时稳定性、使用稳定性、运输稳定性,是贯穿于整个试剂研发、上市前评价及上市后监测的重要内容,是产品有效期设计的依据。 体外诊断试剂在储存、运输及整个使用过程中,各种影响因素如温度、湿度、光照、反复冻融、开瓶、振动等均可能对产品稳定性造成影响。稳定性研究周期一般较长(6至24个月),在研究设计之初充分考虑产品自身特性、对研究条件进行优化设计可避免时间浪费、降低企业研发成本。 体外诊断书以及稳定性研究中常见问题如下: 1.储存条件未包含最差条件 部分企业设置的温度条件跨度较大,但未进行最差条件的验证。如生产企业应明确声称温度的具体范围如10-30℃,研究过程应包含30℃下的长期研究。 2.研究时间不足 出于节约成本的考量或设计问题,部分企业存在未在T0(开始点)进行研究、中间时间点过少、未涵盖声称有效期后时间点等问题。应注意开始时间点应尽量接近产品生产时间,保留基础数据以利于与后续研究结果的比对;增加中间时间点的设置,收集更多的数据点有利于更早识别数据漂移;涵盖预期声明后合理时间,为不确定性预留空间。 3.研究结果不明确 研究结果的不明确源于接受标准的设置模糊,如精密度参考品未明确CV要求、质控品未设置数据偏倚的可接受水平而以“在靶值范围内”作为接受标准。基于原始检测数值和明确的可接受标准,生产企业应明确数据处理过程,可以表格、图示等形式展示产品的质量变化趋势,根据结果分析确定有效期声称。 定性体外诊断试剂阳性判断值变化时是否需要提交检验报告? 需要。定性检测试剂阳性判断值发生变化时可能导至样本的检测结果发生改变,无论产品技术要求中的性能指标及检验方法是否发生变化均应提交检验报告,可为委托检验报告或自检报告。 体外诊断试剂产品技术要求中质控品及校准品的性能指标是否需要考虑均一性? 对于单独注册的质控品或校准品、包含质控品和/或校准品且能报告具体量值的体外诊断试剂,技术要求中性能指标应至少包括质控品和/或校准品的均一性指标,其他指标可依据产品具体情况进行设置。 已上市体外诊断设备仅变更生产地址,是否需要提交分析性能评估资料? 国产医疗器械生产地址变更的,注册人应当在获得相应的生产许可后办理变更备案。进口医疗器械的生产地址发生变更的,需要办理变更注册,提交新的企业资格证明文件,不需提交分析性能评估资料。 如何选择体外诊断试剂的参考区间确定的路径? 根据《体外诊断试剂参考区间确定注册审查指导原则》,确定体外诊断试剂的参考区间可通过参考区间的建立和验证两种方式完成。申请人或注册人应根据申报产品的适用性选择合适方式研究。所有申报产品均可采用建立的方式确定其参考区间。具体方法参照指导原则要求。 当申报产品同时满足以下条件时,也可采用验证原始参考区间的方法确定其参考区间:第一,原始参考区间的研究应系统全面,具有可信性,如已发布实施的临床检测参考区间标准等;第二,检测系统需具有可比性;第三,参考区间研究的分析前因素需具有可比性,如参考个体的状态,标本采集和处理程序等;第四,参考人群具有适宜性,即原始参考区间研究所采用的参考人群应在人群分布的地理位置、人口统计学特征等方面与申报试剂预期适用人群相一致,或包含符合参考区间建立参考样本数量要求的上述参考人群。考虑到国内业界公认参考区间如卫生行业标准中明确的参考区间,其建立时均采用了广泛适宜的参考人群、规范的分析前因素等,因此申报产品在选择该参考区间进行验证前,可不再考虑第三和第四条。 对于质控品稳定性研究,是否需要在稳定性研究进行均匀性评价? 质控品稳定性主要考察质控品量值随时间变化的趋势,一般情况下无需进行均匀性评价。 体外诊断试剂注册变更时,什么情况下需要提交产品检验报告? 体外诊断试剂注册变更时,如涉及产品技术要求变化,如性能指标、检验方法、企业参考品、国家参考品变化,应提交针对产品技术要求变化部分的检验报告。可以为自检报告或委托检验报告。一般情况下,产品技术要求不发生变化的,无需提交检验报告,但当产品发生反应体系、阳性判断值等可能影响检验方法、结果判断的实际变化时,应重新考虑检验用产品规格的代表性,必要时提交新的检验报告。 进口体外诊断试剂是否要按照国内说明书编写指导原则提交中文说明书? 是的。在中国进行注册的体外诊断试剂均应按照《体外诊断试剂说明书编写指导原则》要求编制中文说明书。对于原产国说明书较为简单,缺少相应内容的情况,应根据上市文件、研究资料、中国境内临床试验等内容,完善中文说明书。 临床试验的比对研究,是否可以只选择1种比对方法,如金标准培养法与对比试剂选择其中1种? 对比方法的选择需要结合申报产品的具体情况确定。 例如病原体相关标志物检测用于病原体感染辅助诊断的产品: 如果有已上市同类产品,可以选择与已上市同类产品进行对比试验;如为全新产品、尚无同类产品上市的情形,一般需分别与检测性能具有较好可比性的实验室参考方法和病原体分离培养鉴定方法进行比较研究,需要时还应与目标疾病的临床诊断结论进行比较研究。如有相关指导原则应参照指导原则的要求执行。 CAIVD综合整理 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号