金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
本文首发于微信公众号 BioArtReports
BioArt,一心关注生命科学,只分享更多有种、有趣、有料的信息。
关注请微信搜索公众号bioartreports。投稿、合作、转载授权事宜请联系微信ID:fullbellies
或邮箱:sinobioart@bioart.com.cn
撰文丨奚望
责编丨迦溆
中国是一个拥有14亿人口的多民族国家,针对全体中国人的群体遗传学研究具有重大的科学价值。然而由于基因测序成本和样本数量的限制,中国人口的群体遗传特征并未得到过充分的研究。
10月4日,深圳华大基因(BGI)的研究人员(徐迅、汪健等)与丹麦哥本哈根大学以及加州大学伯克利分校的研究人员合作在Cell期刊上发表了名为“Genomic Analyses from Non-invasive Prenatal Testing Reveal Genetic Associations, Patterns of Viral Infections, and Chinese Population History”的文章,借助非侵入性产前检测(NIPT)技术对超过14万来自中国各地的女性进行了基因组测序和分析。他们利用这份大规模数据揭示了中国人遗传历史与地理因素的关联,并发现了多个在中国人血液中富集的病毒类型。

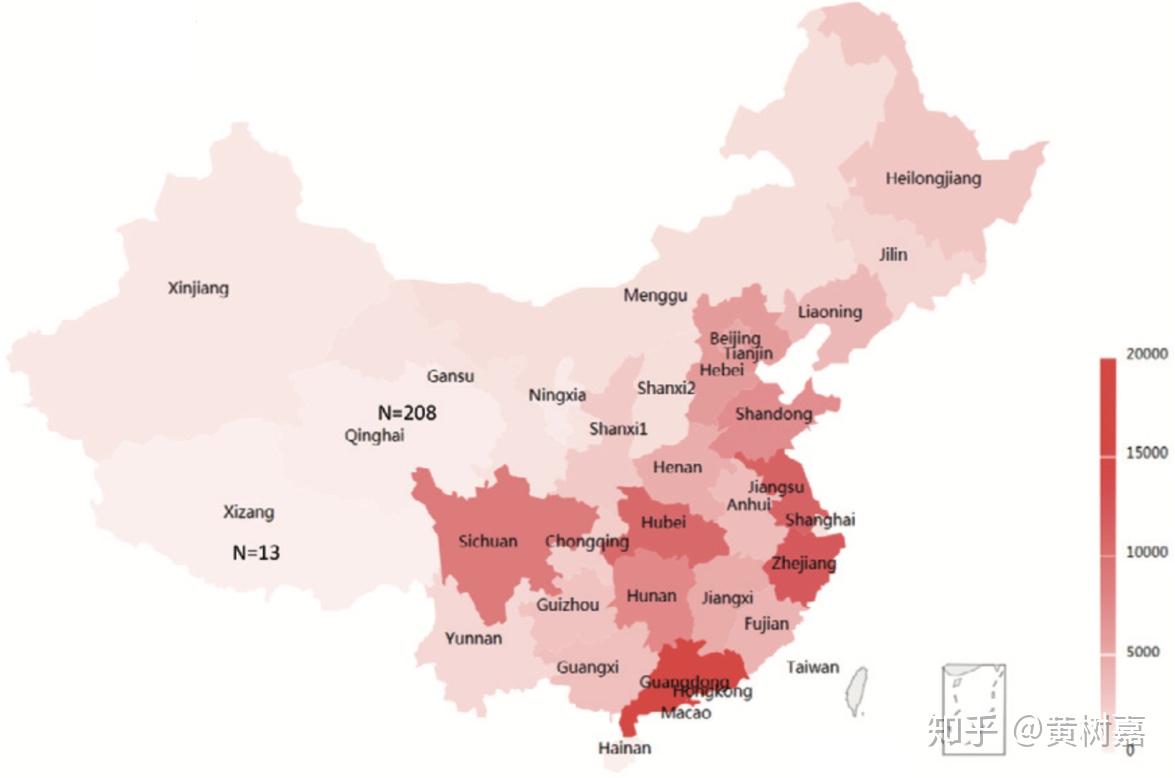
非侵入性产前检测(NIPT)的目的,是通过提取怀孕母亲血液中的游离DNA对胎儿进行21三体综合征的筛查。世界范围内已有上千万女性接受过这项检测,其中6-7百万来自中国。虽然样本量巨大,但NIPT的测序深度通常只有0.06X-0.1X,与一般的全基因组测序(15X-30X)相差巨大。不过在相关研究的佐证下【1】,研究人员通过统计手段对基因型数据进行估计与填补,分析了来自中国31个省市自治区,36个民族女性的测序数据(由于华大基因拥有海量的NIPT的数据,所以大样本就不是问题了)。他们从中发现了约9百万可靠的单核苷酸突变位点,其中有超过20万不存在于已有的数据库中。
研究人员对所有样本的基因型进行了主成分分析(PCA),他们发现在汉族人口中,人们居住地纬度的不同伴随着显著的基因差异,而东西部人口间的差异则并不明显。这可能是因为我国存在大量人口向西部迁移造成的。相比汉族内部,各少数民族与汉族间的基因型差异更为明显,其中差别最大的包括来自新疆的维吾尔族、哈萨克族和来自内蒙古的蒙古族。来自中部的回族,西南的彝族和南方的壮族、布依族携带的基因变异也与汉族非常不同。而由于历史原因,满族人的遗传信息则和东北方汉族人接近。
作者接着比较了汉族和世界其它主要民族的基因相似度。他们发现西北和中西部中国人同欧洲人(CEU)较为相似,其中相似度最高的来自新疆、甘肃、青海和宁夏等地。作者认为这与河西走廊在汉代丝绸之路中扮演的重要地位有关。而新疆、西藏、海南、云南和广西等地的人口基因型同印度人存在相似性。作者还使用统计方法对汉族内部的基因型选择进行了推断。他们发现LILRA3、CR1、 FADS2、 DOCK9、 ABCC11和IGH基因簇伴随居住地纬度的不同存在明显的差异。这些基因同免疫反应,躁郁症和饮食相关。一些ClinVar数据库收录的疾病关联等位基因也呈不同的地域分布。
而后,作者使用全基因组关联(GWAS)分析对基因型和表型间的相关关系进行研究。他们首先选取身高和BMI指数作为目标表型,并分别发现了48个和13个基因组位点与之显著相关,其中有41个和10个都曾被报道过,证明了用低测序深度的大样本数据做关联分析也能得到可靠的结果。随后作者选用了生育年龄和双胞胎怀孕这两个表行来研究受孕与基因型的关联。他们发现了一个在EMB基因附近的位点与生育年龄呈强相关性,而NRG1基因上的一个SNP与双胞胎怀孕的相关性最强。EMB基因在胚胎发育中扮演着重要作用,NRG1基因则主要在甲状腺表达。这两个基因都曾被研究证明与受孕相关。
最后,作者对这些NIPT血液样本中的病毒组成进行了调查。血液病毒研究是病毒流行病学的重要部分。有趣的是,这些中国样本血液中的病毒种类和丰度同欧洲人很不一样。乙肝病毒(HBV)和B19微小病毒在这些样本中丰度很高,人类疱疹病毒7型则含量很少。作者发现基因组上MLC1- MOV10L1区域里的一个SNP同人类疱疹病毒-6A/B的存在具有很强的关联。MOV10L1基因是一个结合PIWI RNA的解旋酶,作者认为对这个基因活性的抑制可能会给人类疱疹病毒-6A/B的侵入提供更佳的环境。同时,血液病毒的分布也呈现出不小的地域差异,如HBV病毒在南方的分布相比北方更为广泛。
参考文献
1. Pasaniuc B, Rohland N, McLaren P J, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies[J].Nature genetics, 2012, 44(6): 631. |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-12-13 22:52
发表于 2024-12-13 22:52



 发表于 2024-12-13 22:55
发表于 2024-12-13 22:55