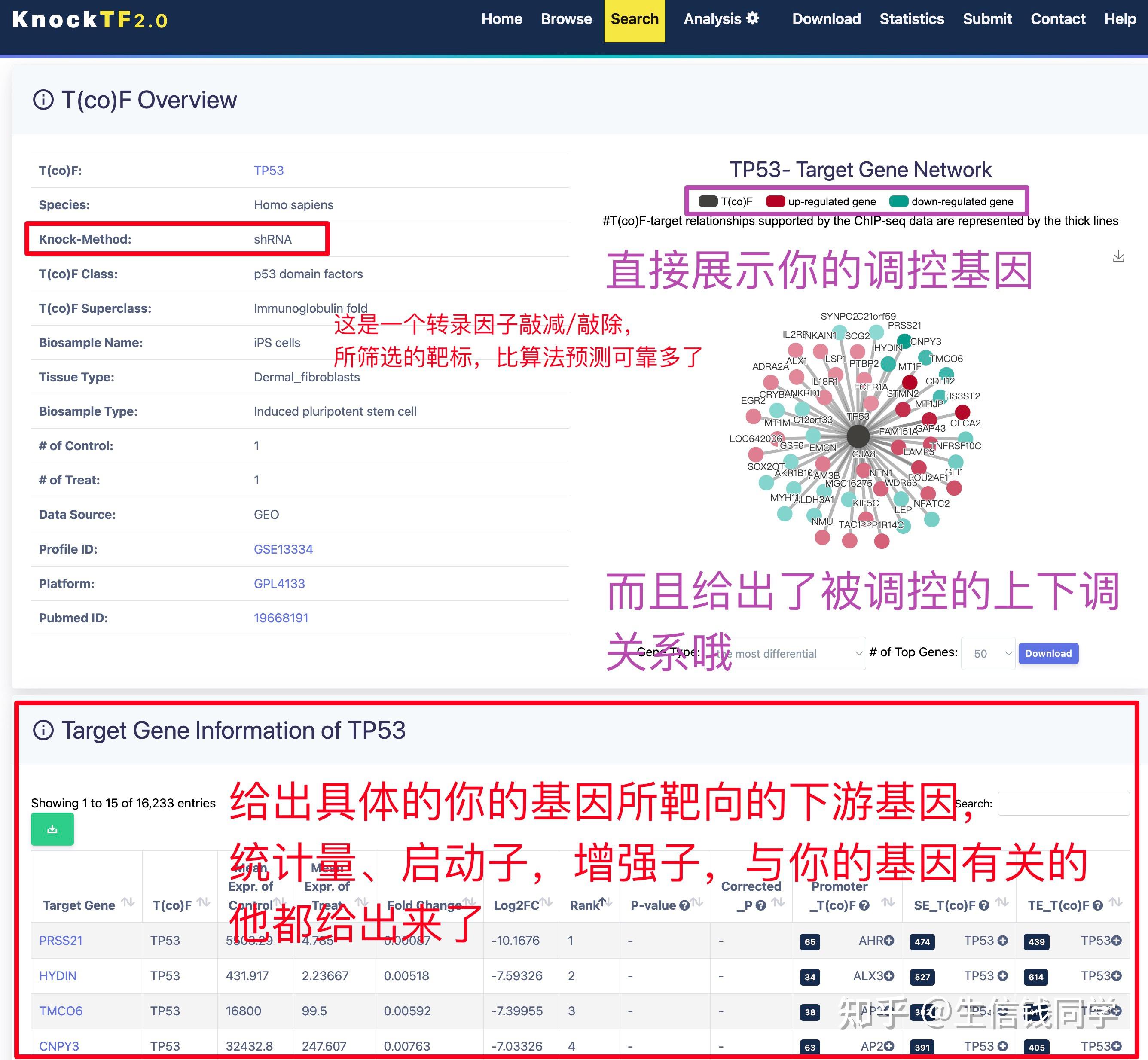
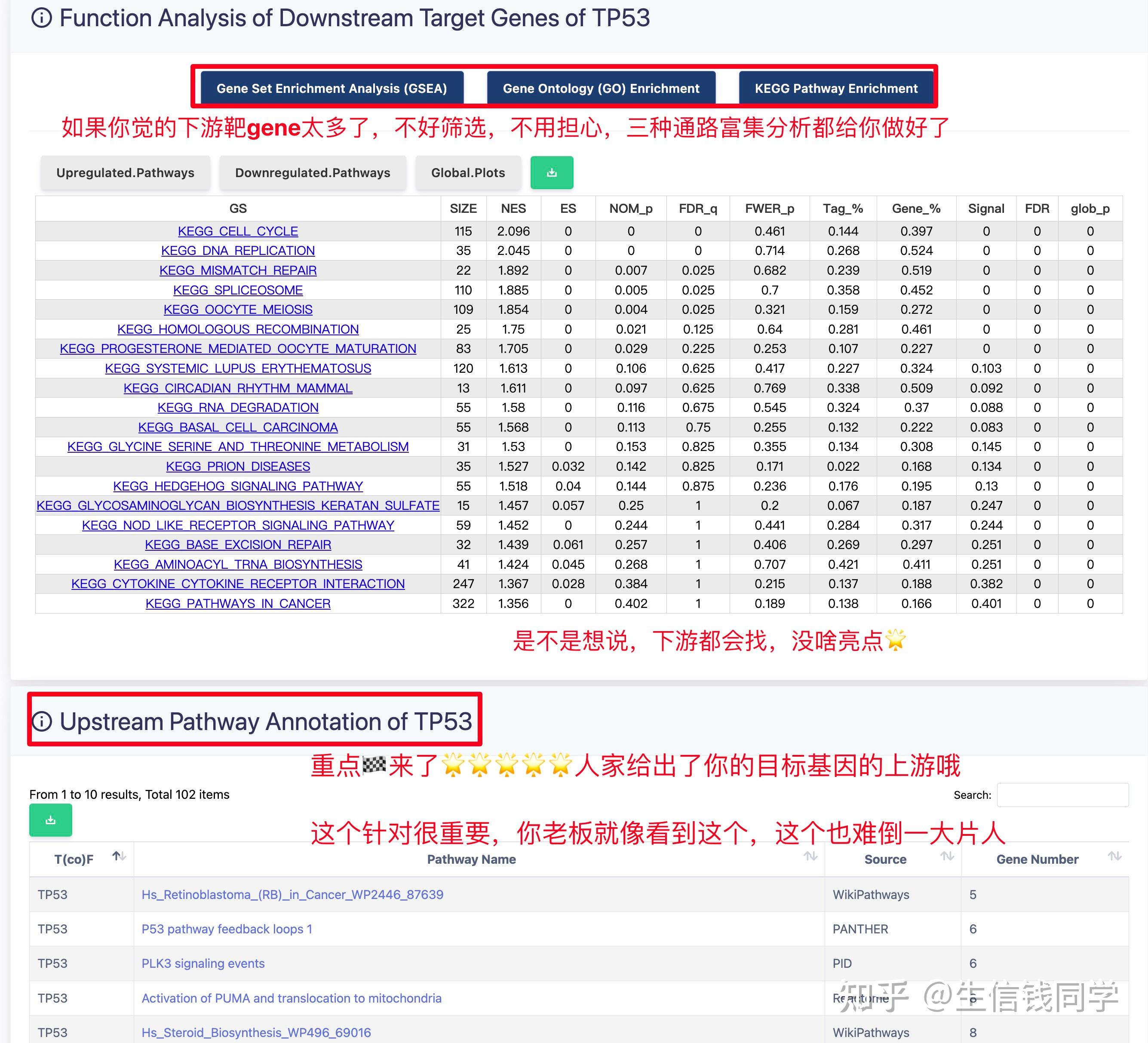
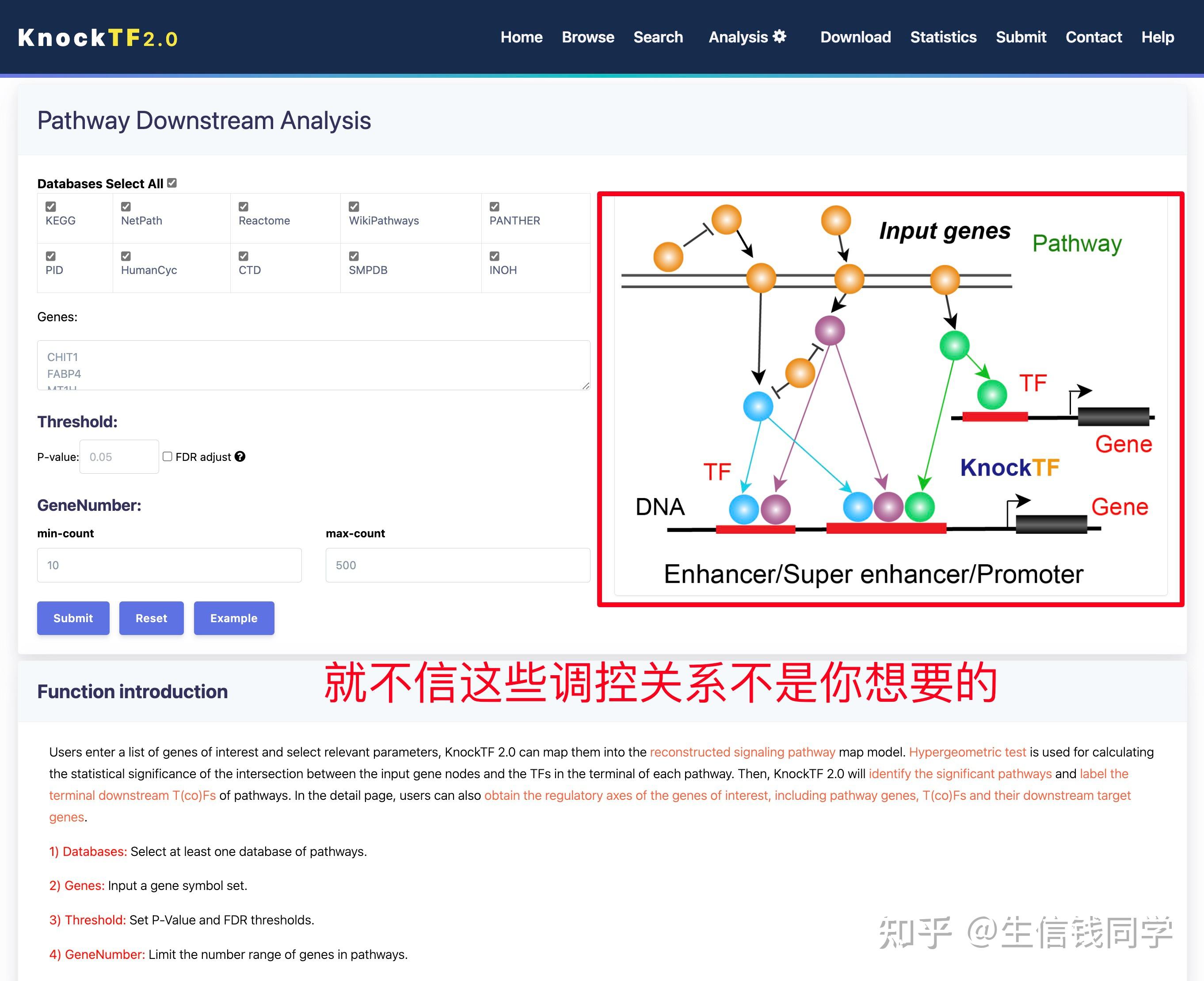
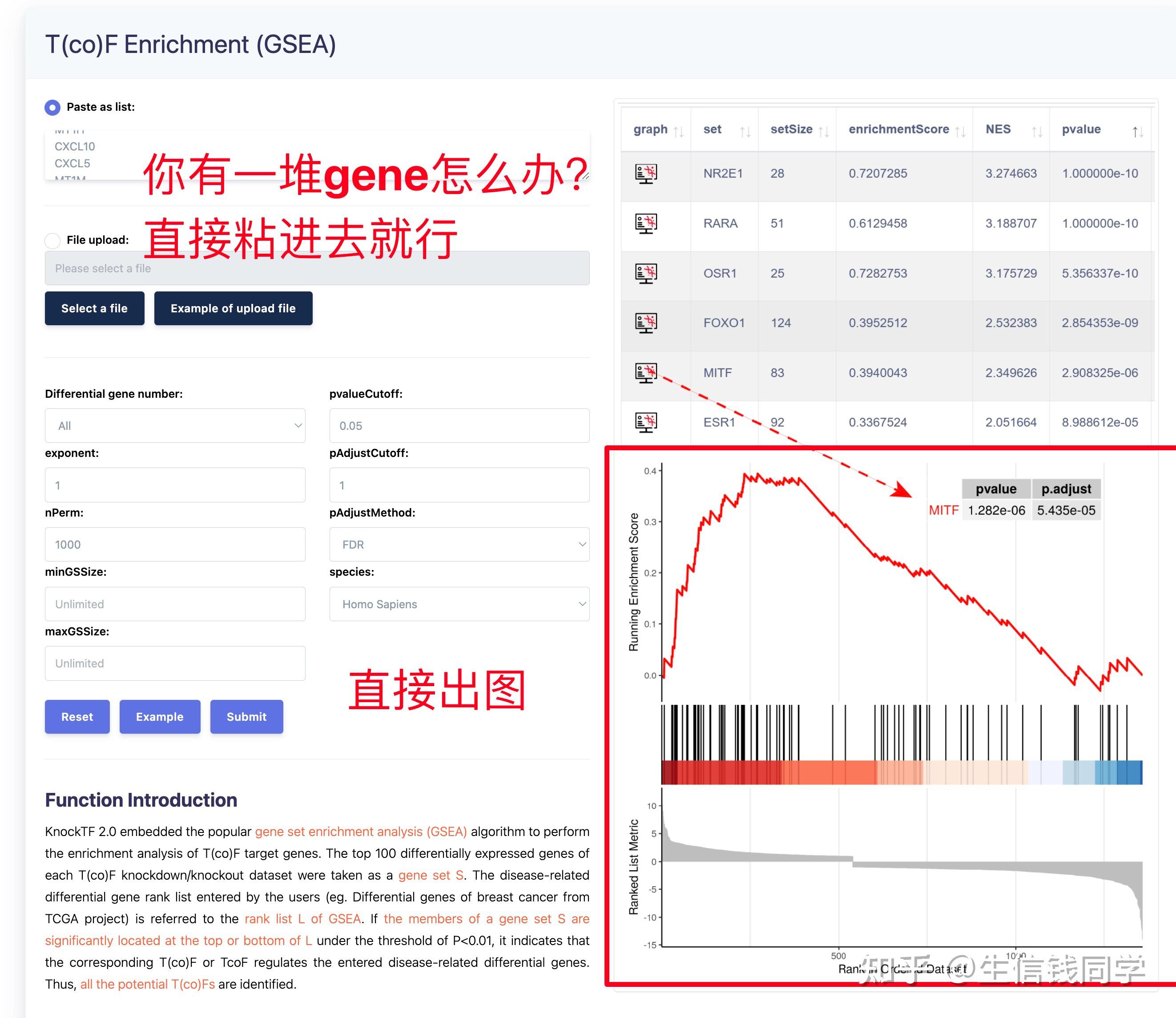
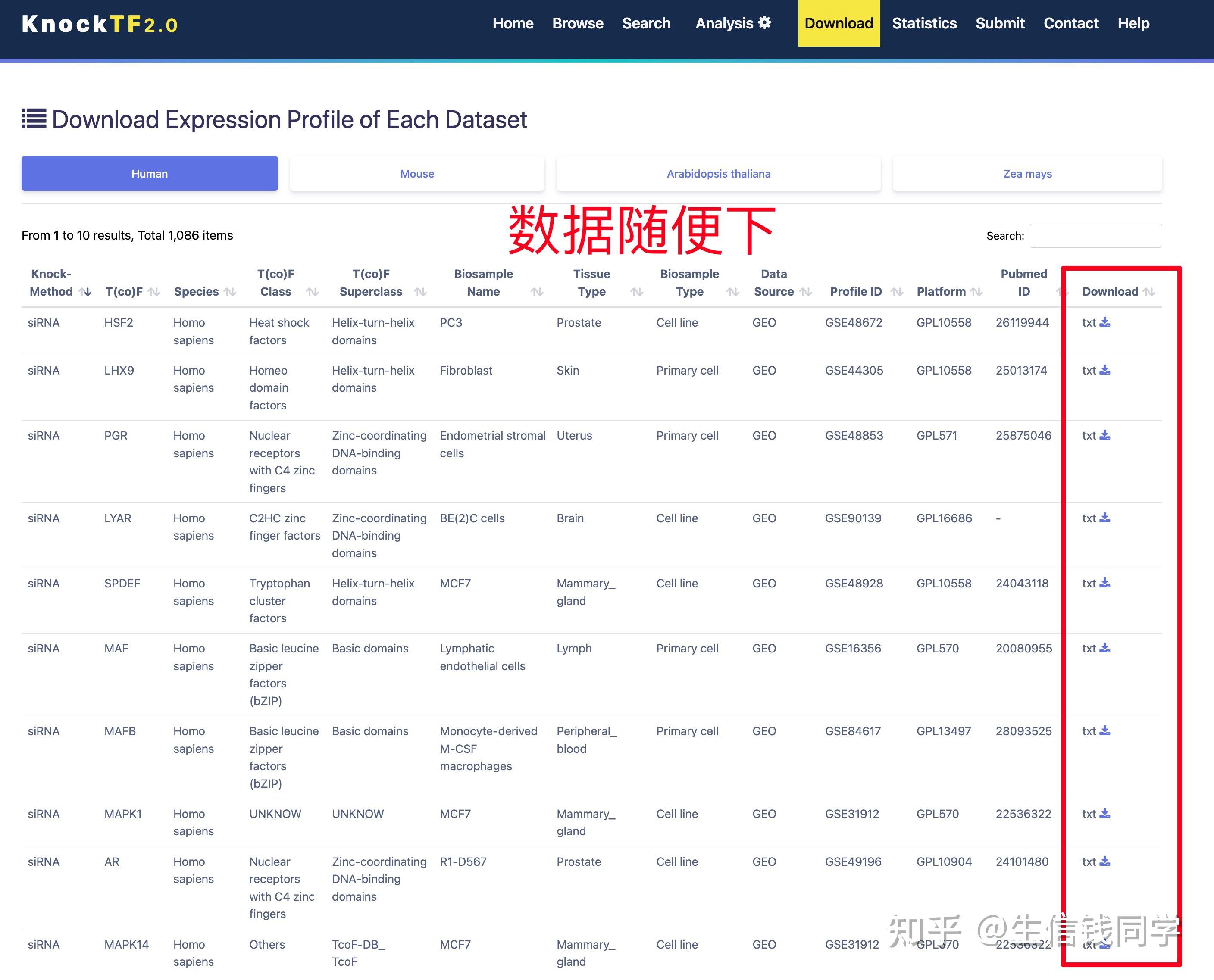
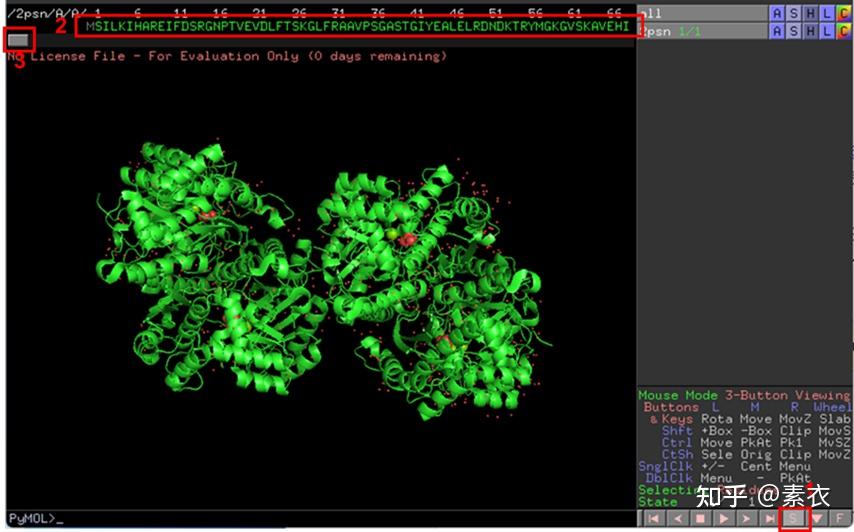
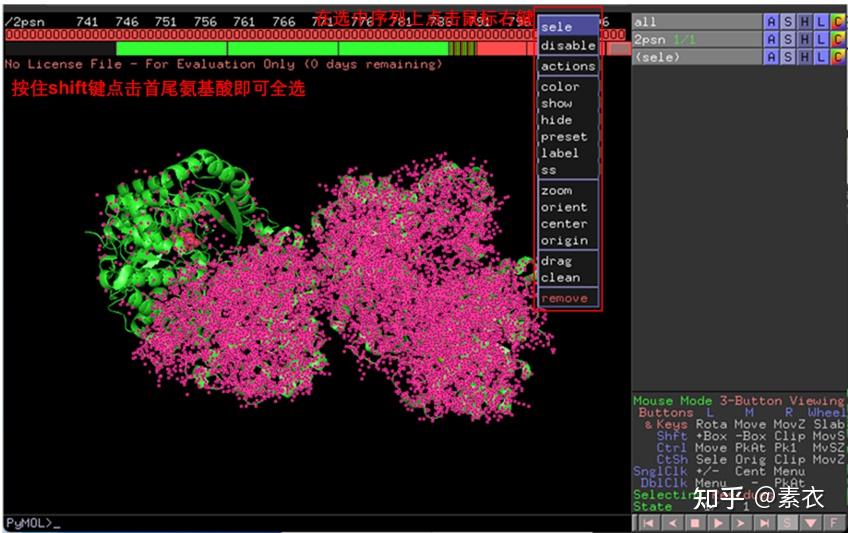
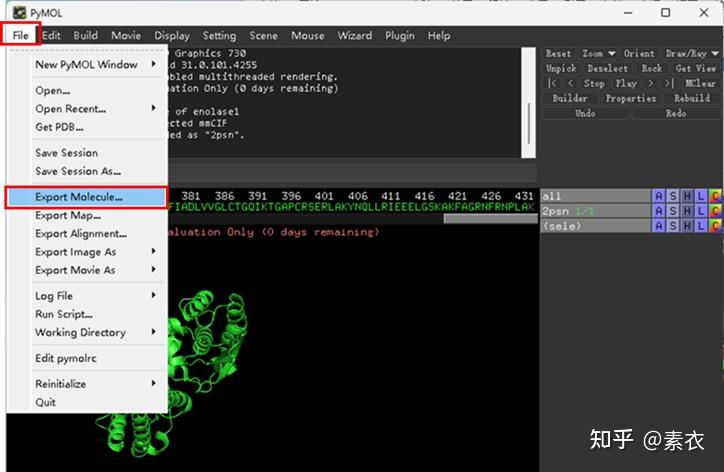
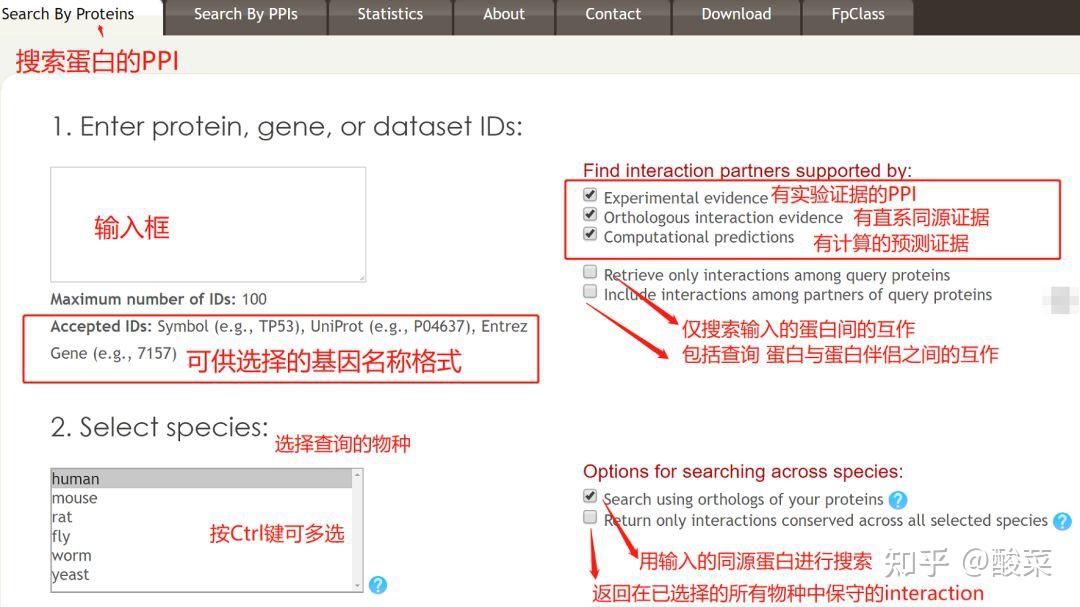
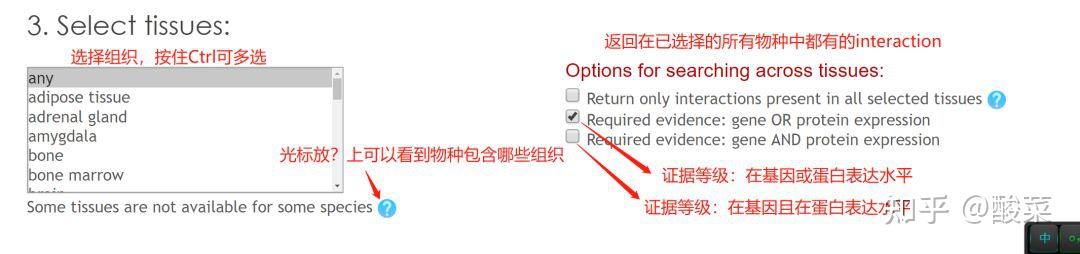
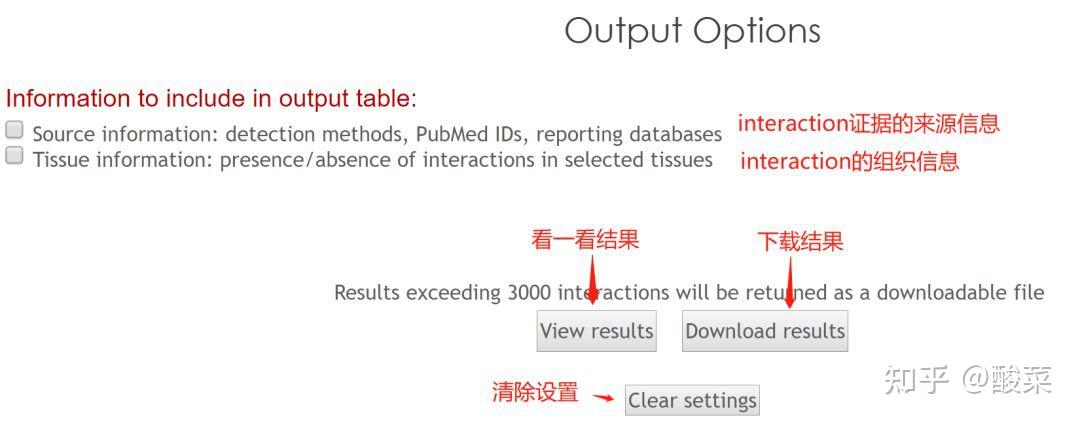
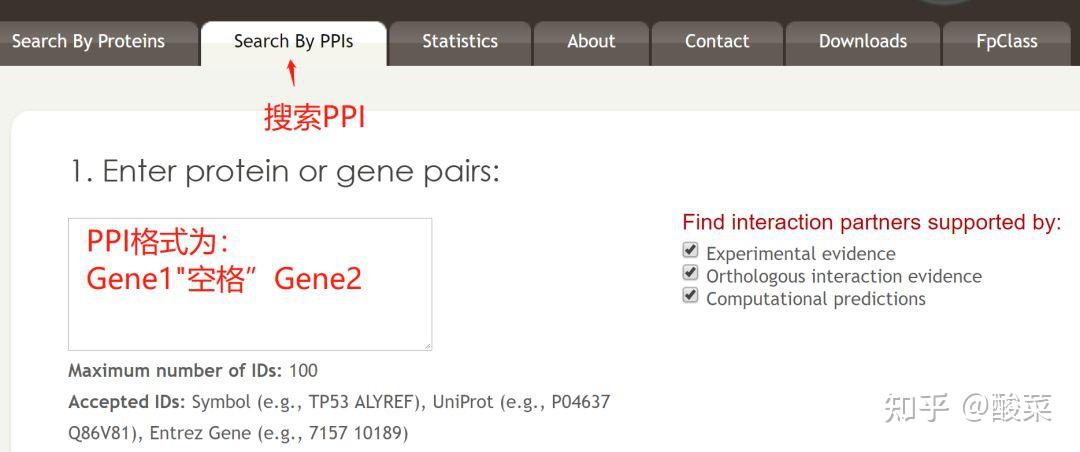
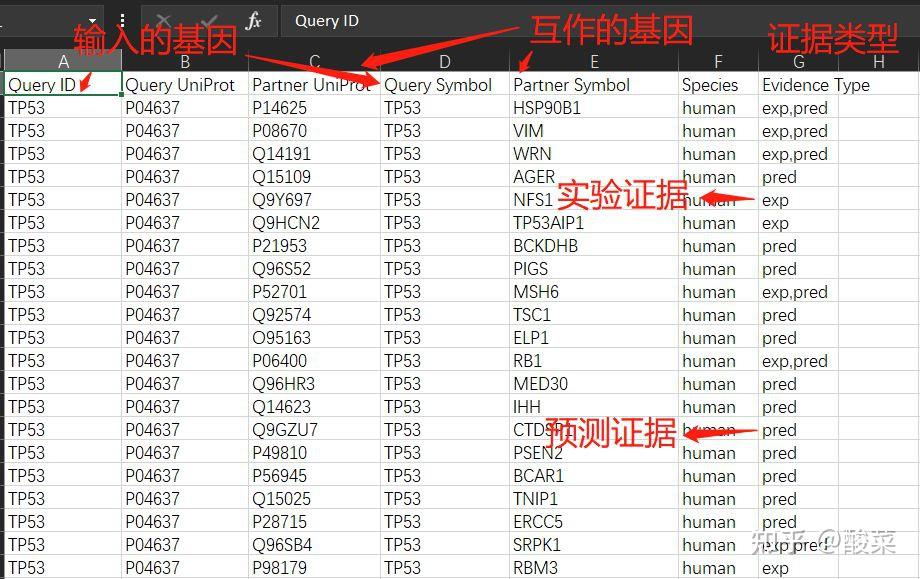
图 3 保存蛋白单体结构文件
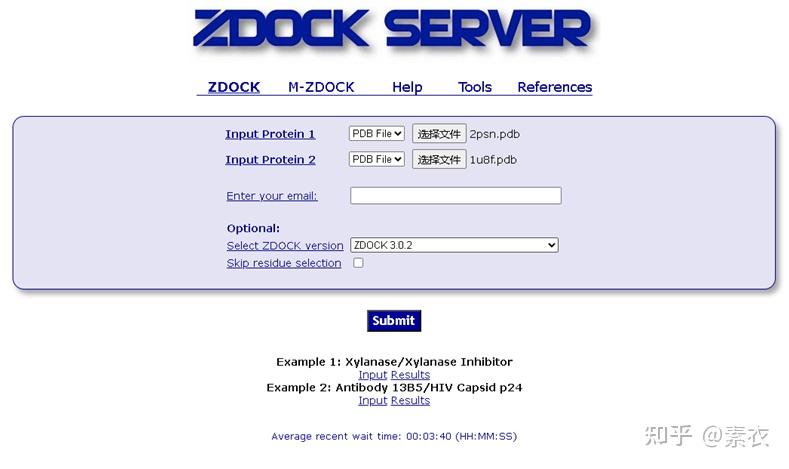
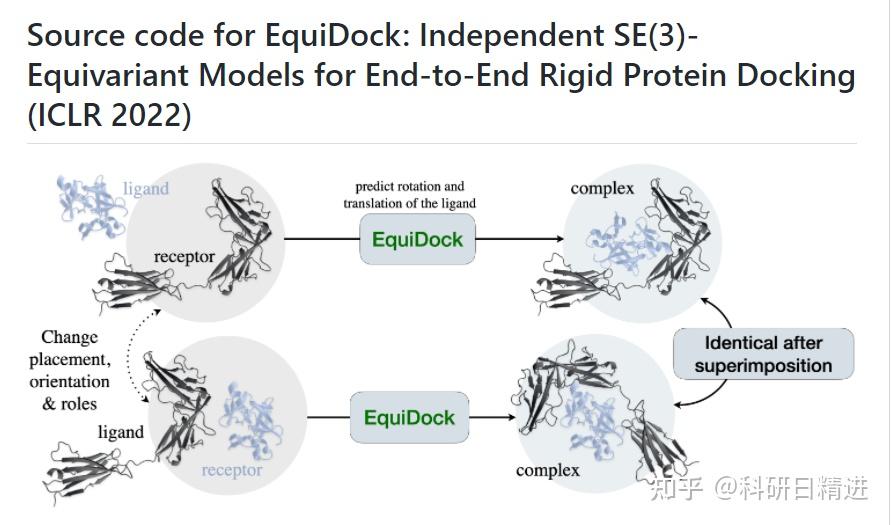
2)ZDOCK蛋白-蛋白对接
进行蛋白质分子对接预测分析工具有很多,常用的包括RosettaDock、HADDOCK、ClusPro、SwamDock等。这里介绍其中一种Z-dock分子对接的使用方法。
ZDOCK网址:ZDOCK Server: An automatic protein docking server (http://umassmed.edu)
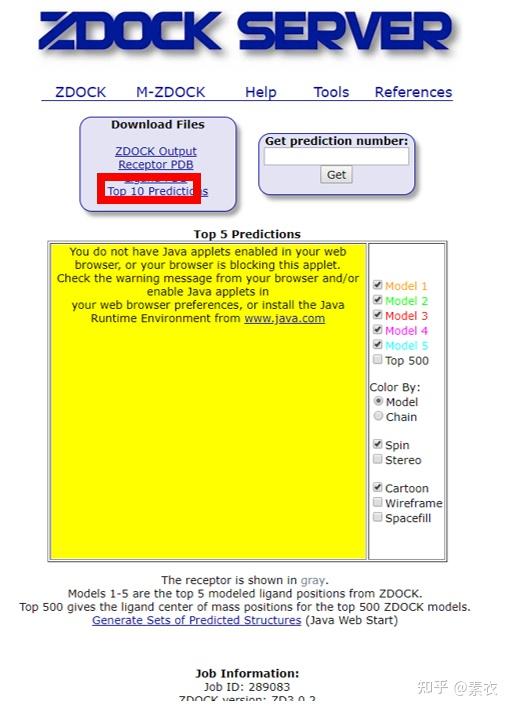
② 通过Z-dock预测蛋白复合体结构;
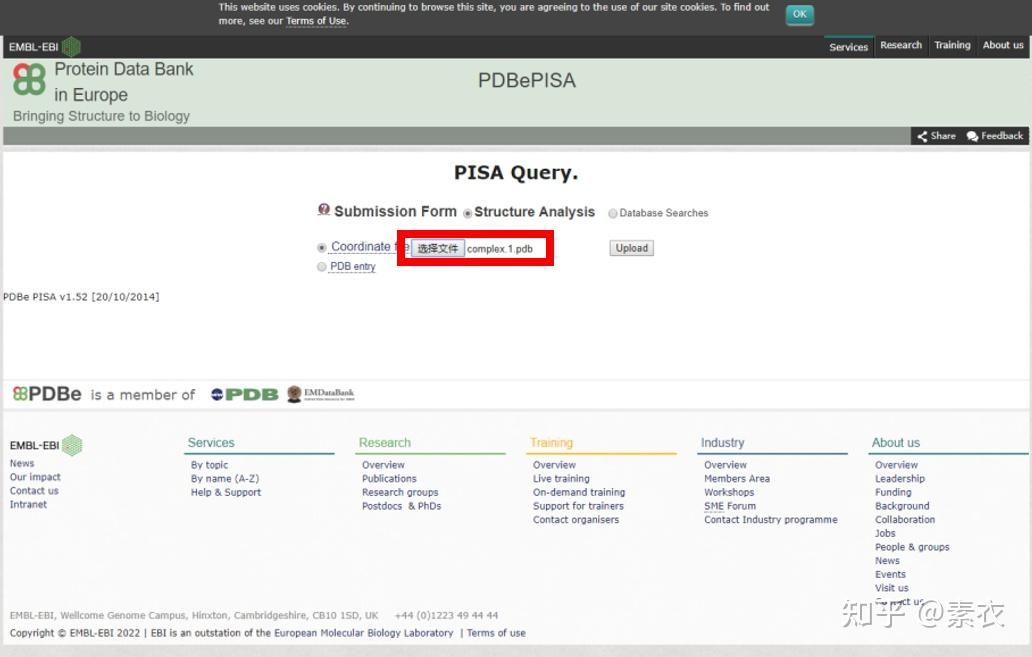
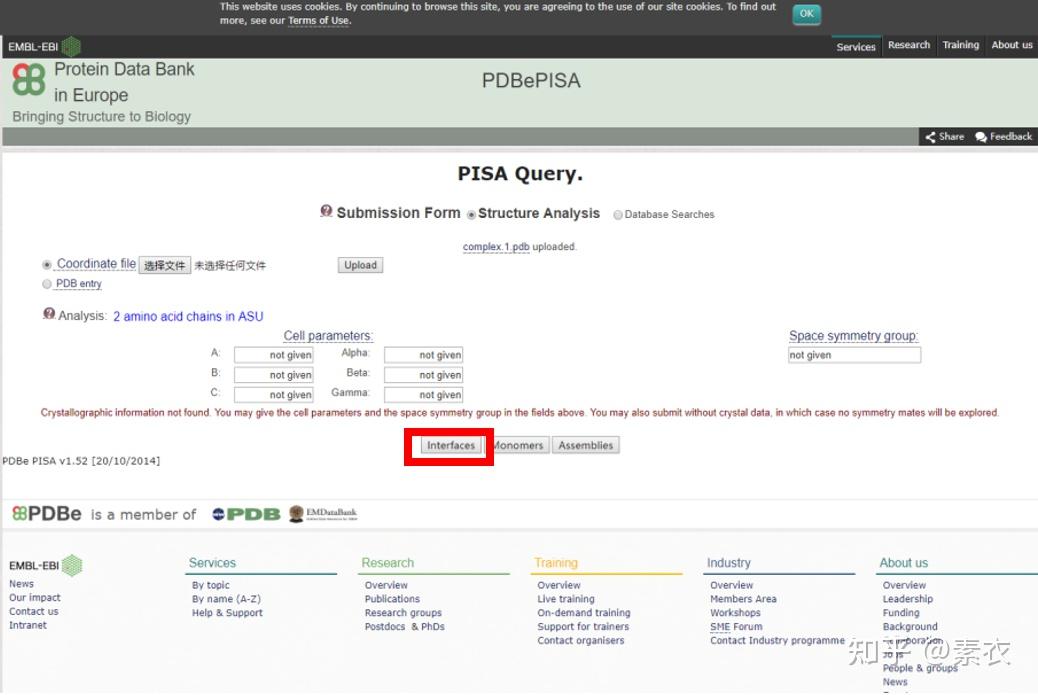
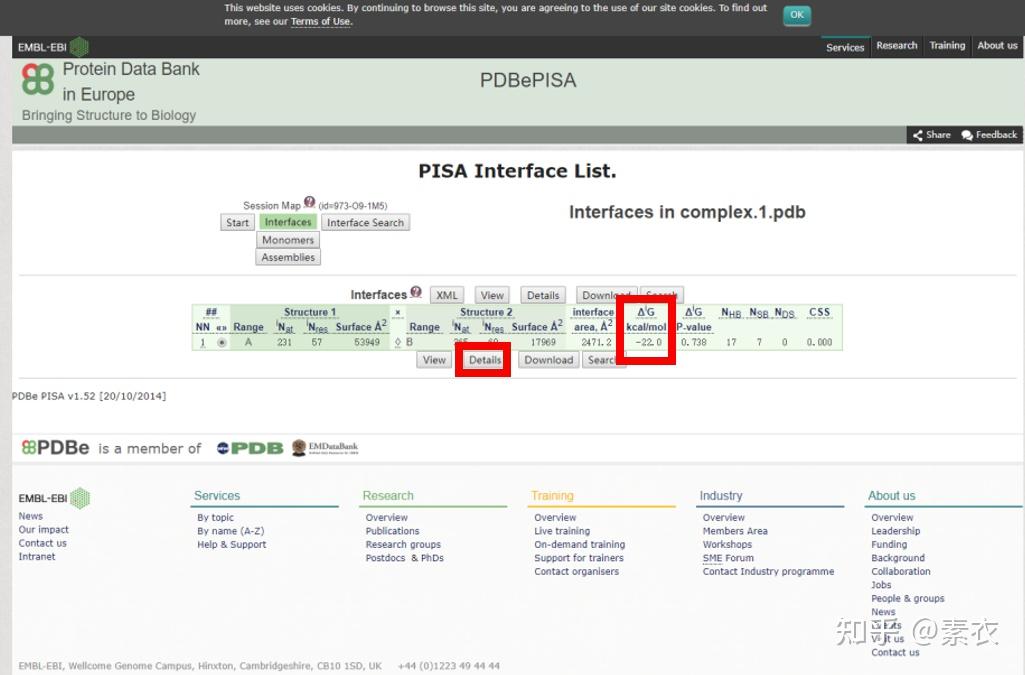
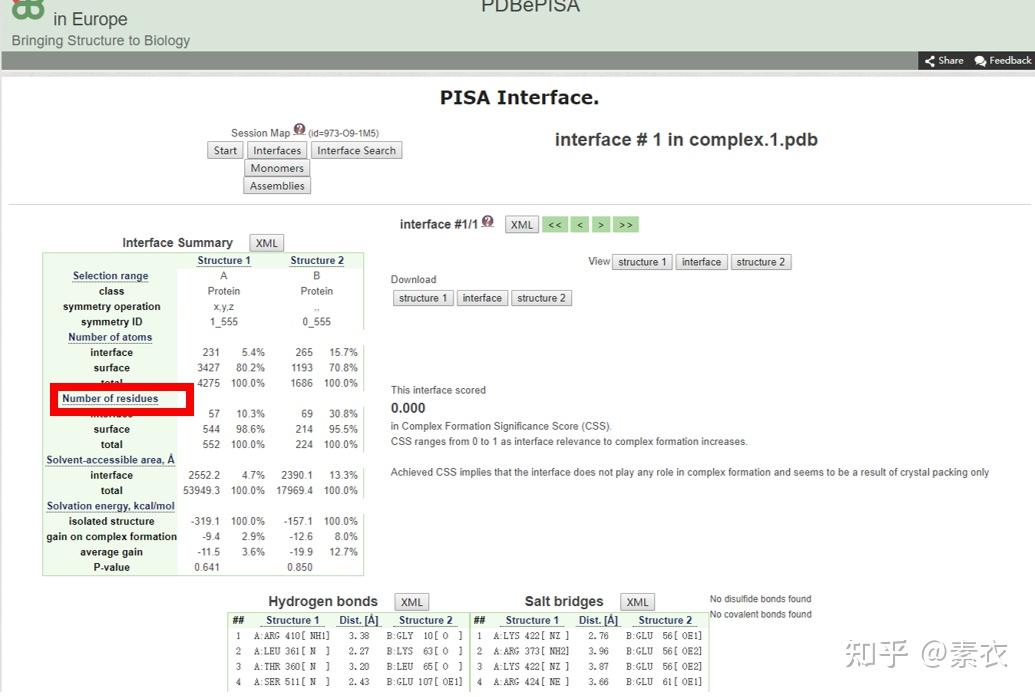
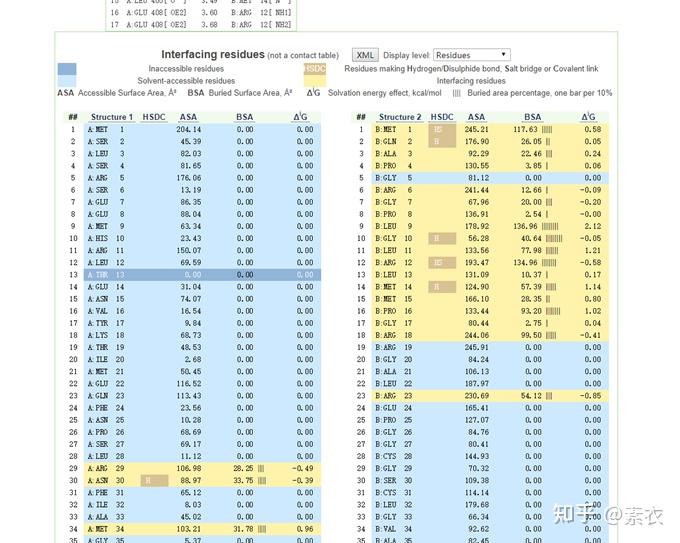
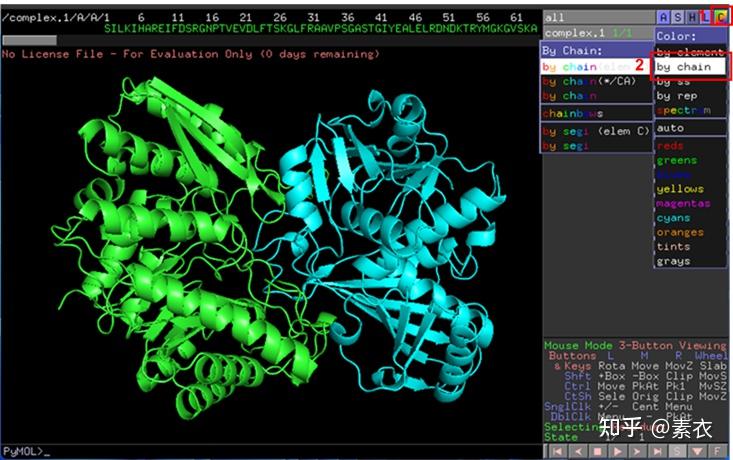
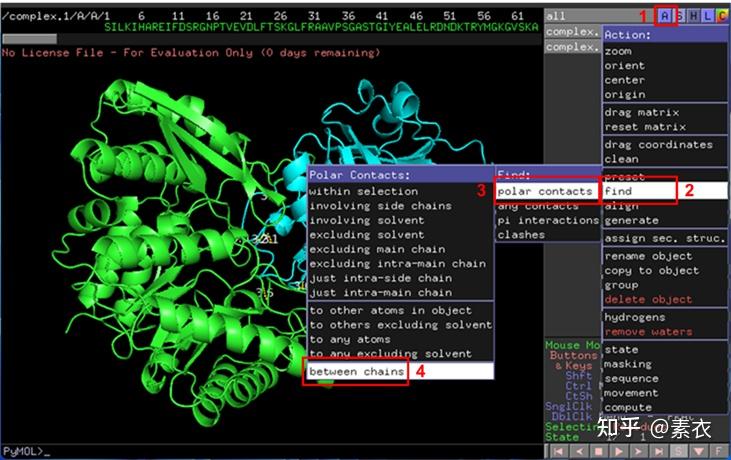
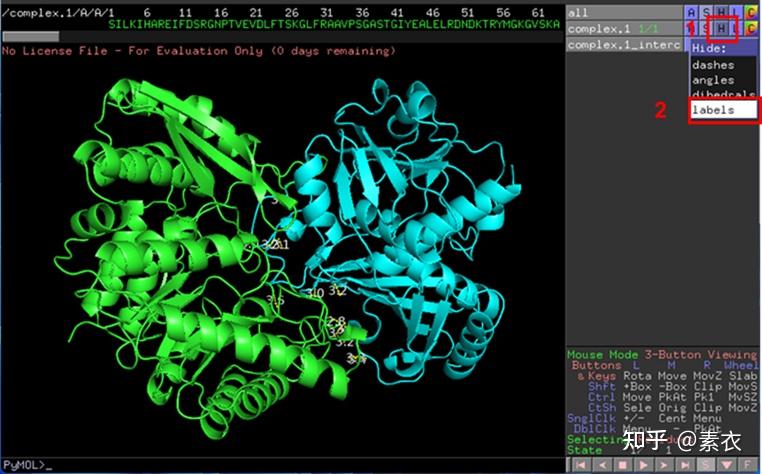
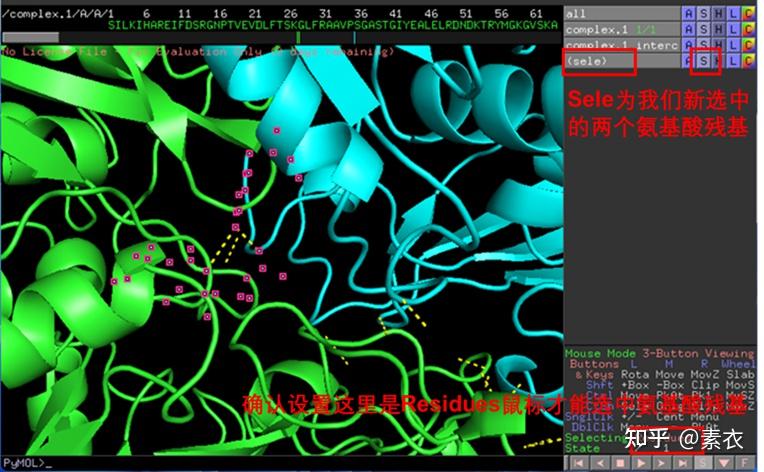
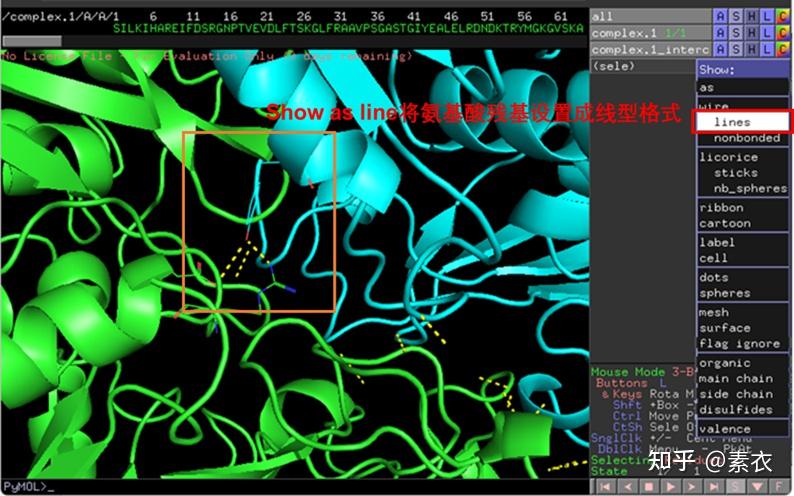
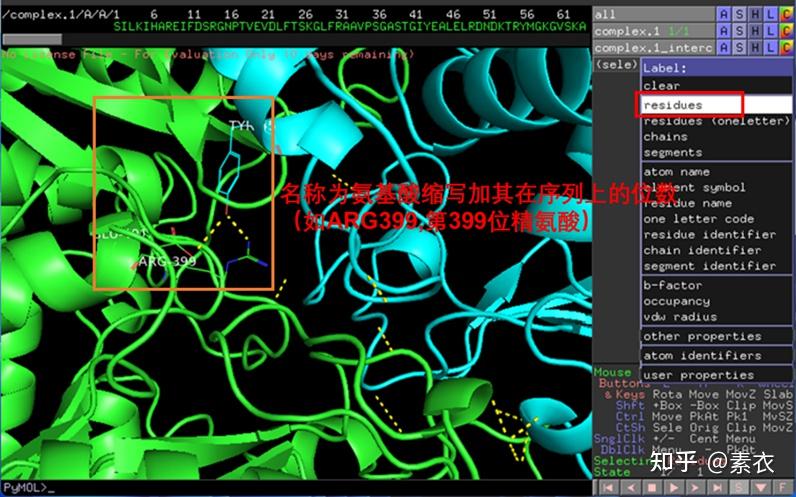
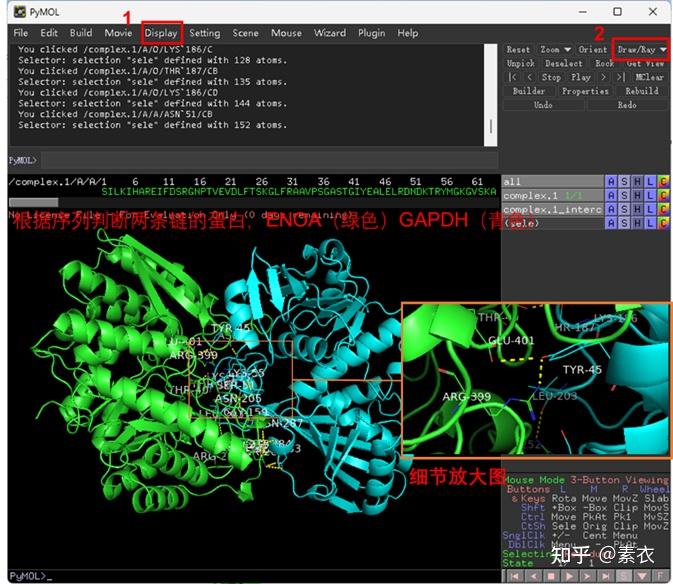
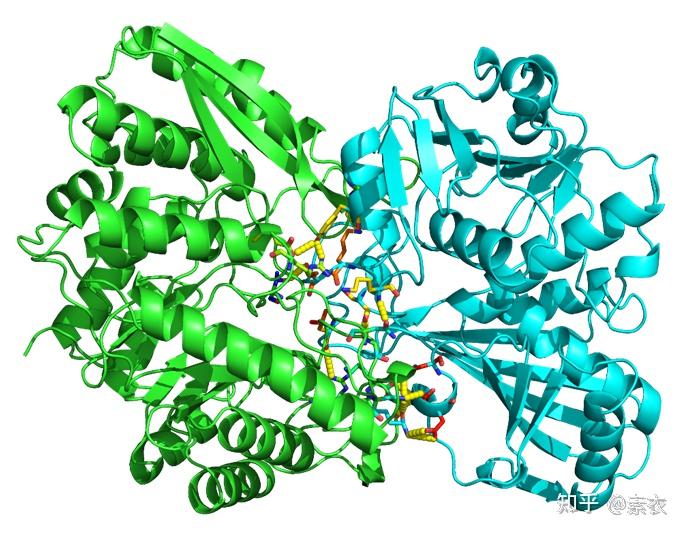
③ 通过PDBePISA分析蛋白复合体中蛋白相互作用的区域,通过Pymol对蛋白相互作用结构域进行可视化分析。
其他问题
1)选择使用PDB数据库下载的蛋白结构还是AlphaFold结构?
如果有完整序列的PDB结构优先使用PDB结构,如果没有可以使用AlphaFold结构。因为PDB结构可能包含水分子或其他离子或配体,还有的是多聚体结构,因此在对接前需要对蛋白结构进行前处理。而AlphaFold结构是纯净的蛋白单体,可以直接使用。
2)蛋白相互作用结构域预测(蛋白对接)可信度如何?
相比而言,小分子-蛋白对接可靠性高,而蛋白-蛋白对接因为蛋白结构较大,而且会有结构柔性变化,因此较难预测。这里所使用ZDOCK是基于FFTs算法的刚性对接,使用较为广泛,对接结果可靠性较高。此外,感兴趣的老师可以关注一下CAPRI-全球蛋白复合物结构预测比赛,里面还会有一些在蛋白相互作用预测中表现好的软件工具。因为蛋白-蛋白对接计算量较大,推荐使用在线服务器平台。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-10-25 22:12
发表于 2024-10-25 22:12


 提升卡
提升卡