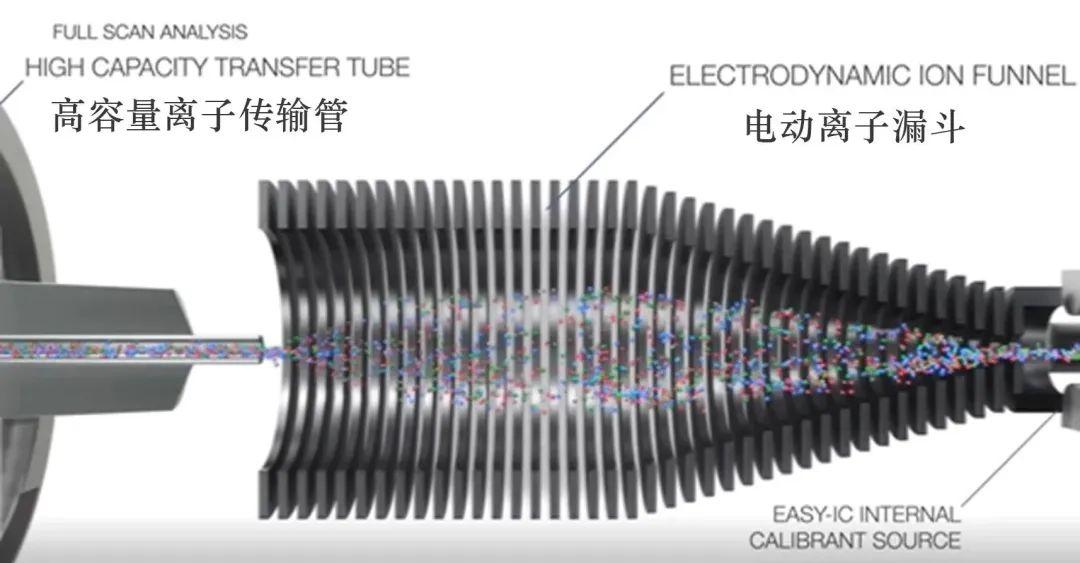
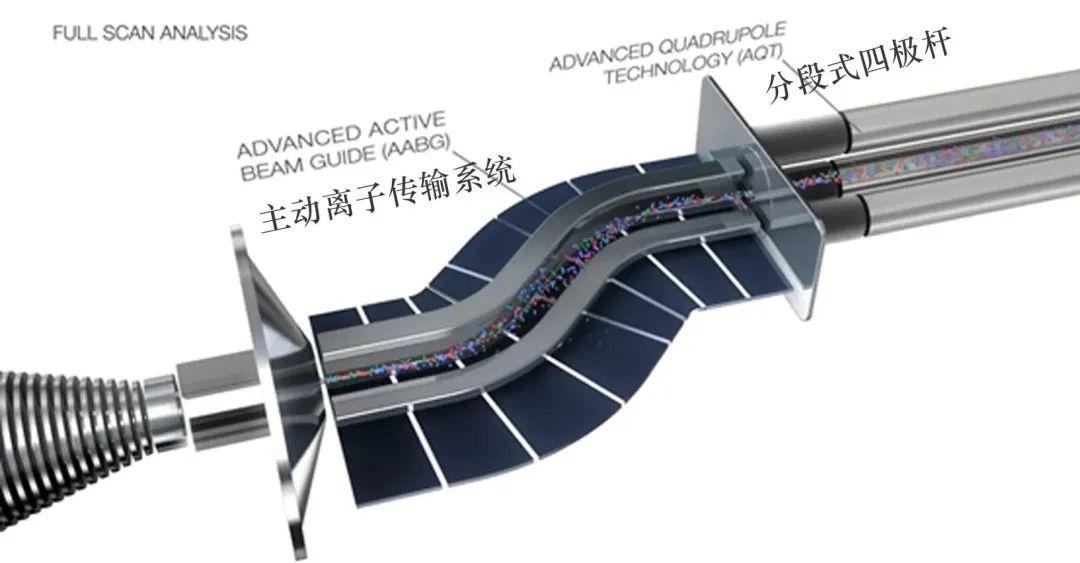
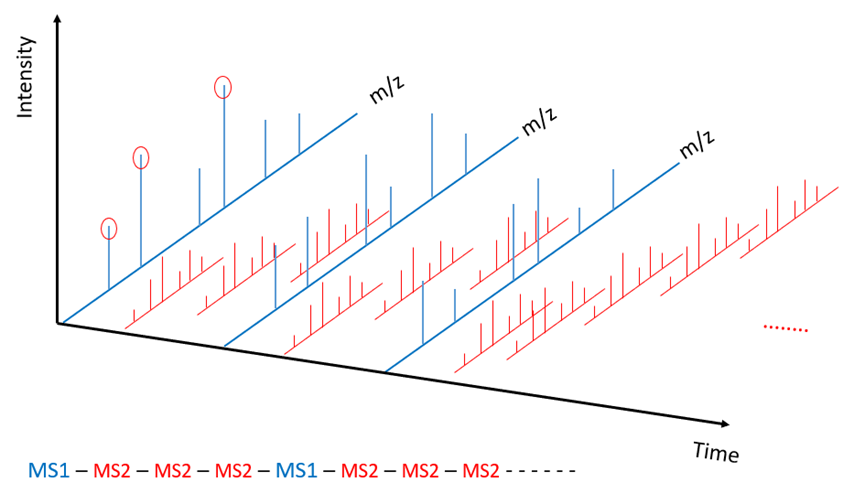
金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
质谱(Mass Spectrometry,MS)是一种强大的分析技术,可用于研究蛋白质的质量、结构、修饰和相互作用等方面。以下是用质谱研究蛋白质的几种主要方法:
1.蛋白质质量分析:质谱可用于测定蛋白质的质量,从而验证蛋白质的纯度和一致性。这种方法通常使用基于矩阵辅助激光解吸/电离(Matrix-Assisted Laser Desorption/Ionization, MALDI)或电喷雾电离(Electrospray Ionization, ESI)的质谱技术。
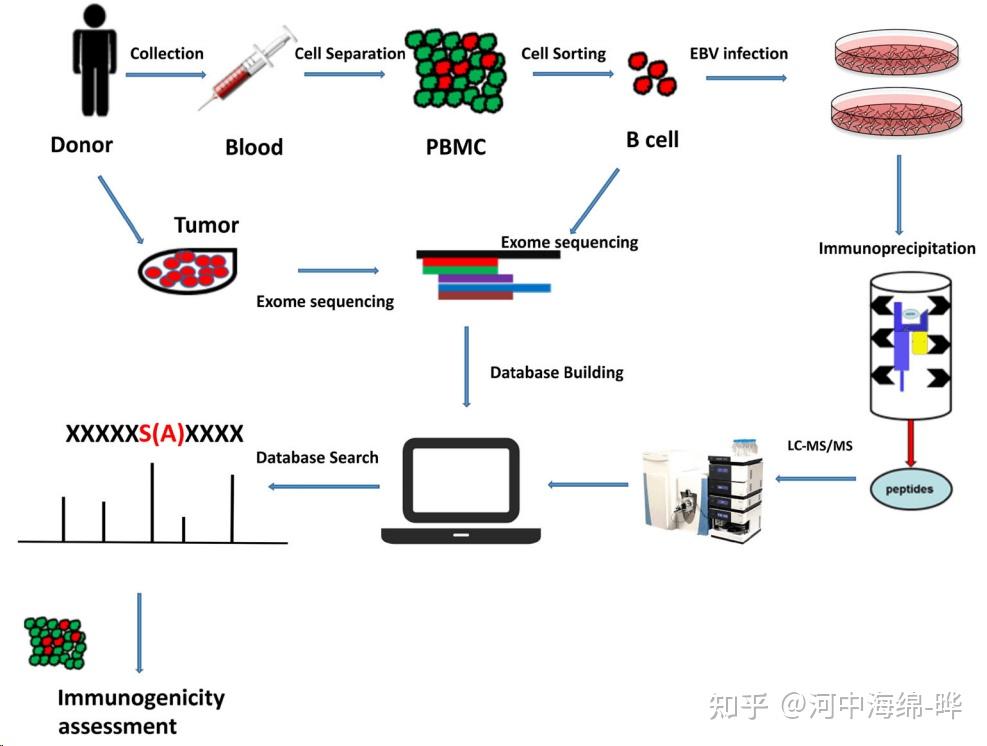
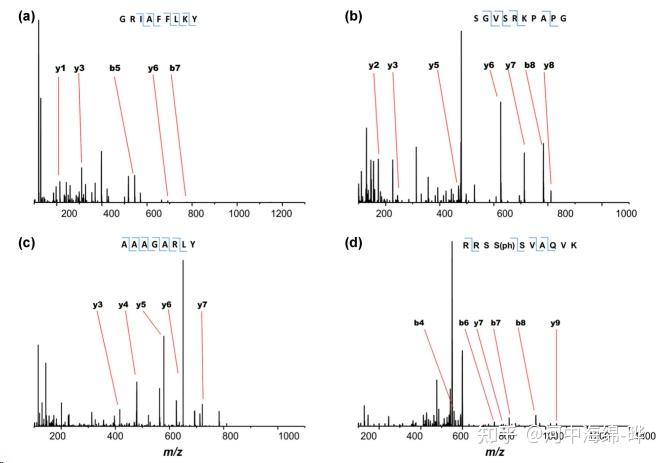
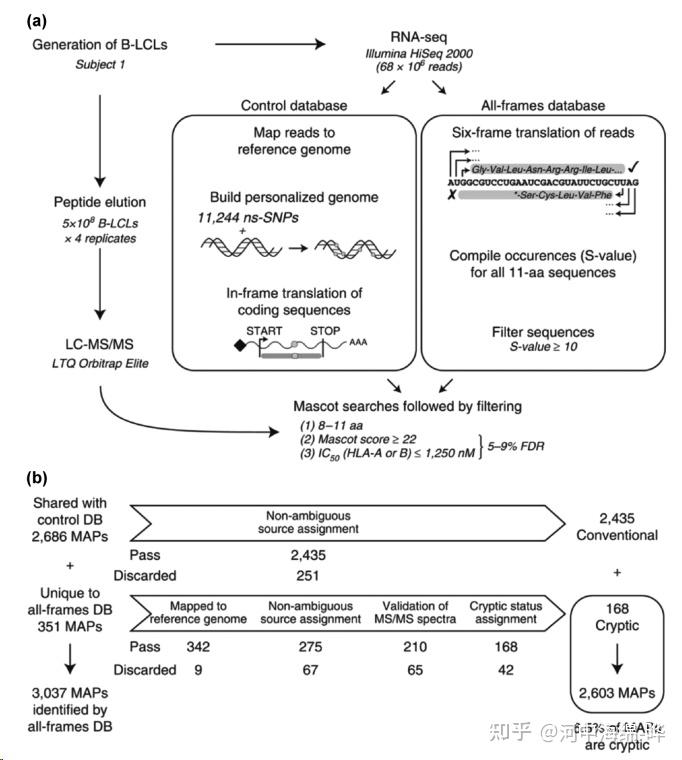
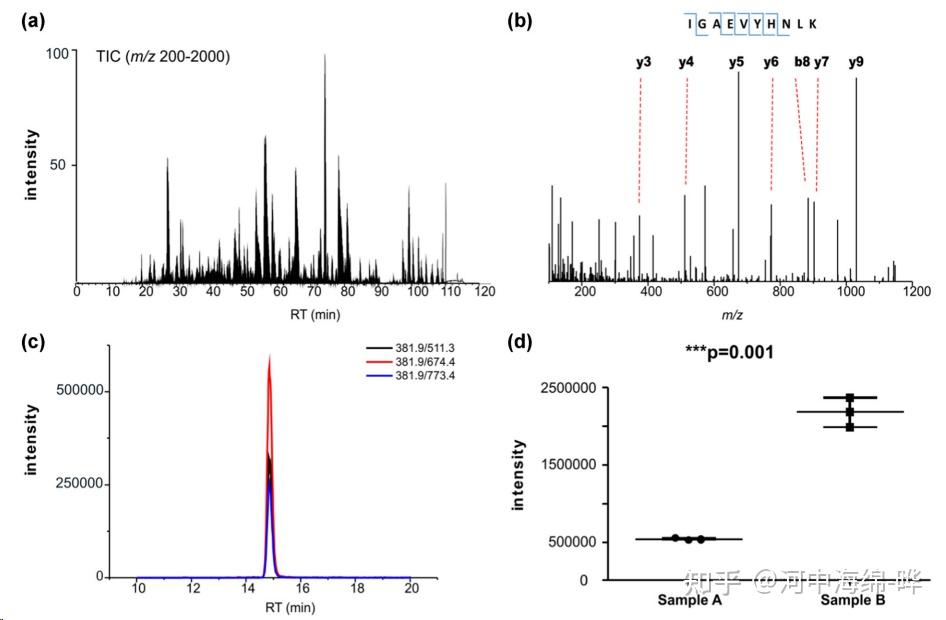
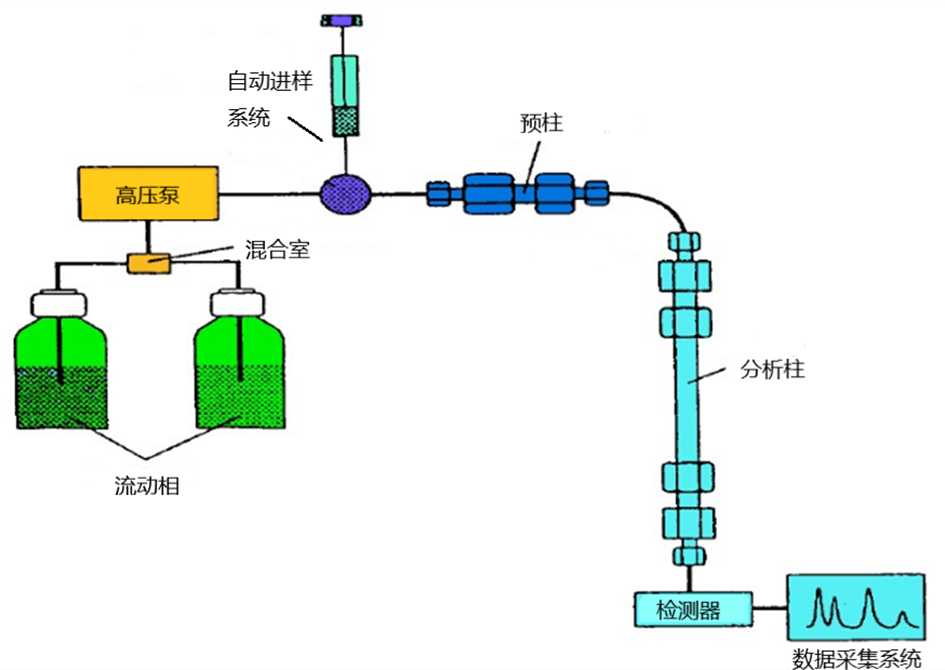
2.蛋白质鉴定和肽段测序:质谱可用于蛋白质鉴定和肽段测序。通过对蛋白质进行酶切(如Trypsin酶切),产生肽段。然后使用液相色谱(如高效液相色谱,HPLC)对肽段进行分离,再用质谱进行检测。最后,通过比对数据库,鉴定蛋白质的序列和来源。这种方法通常使用串联质谱(Tandem Mass Spectrometry, MS/MS)技术。
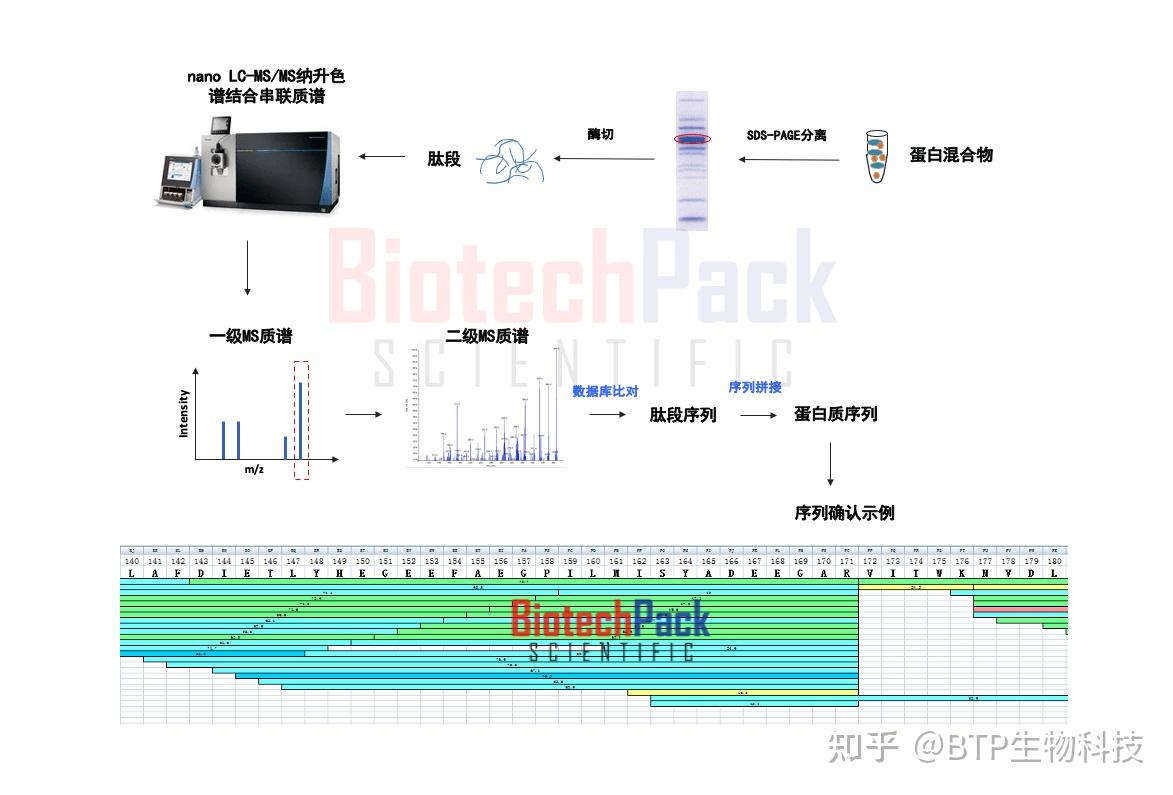
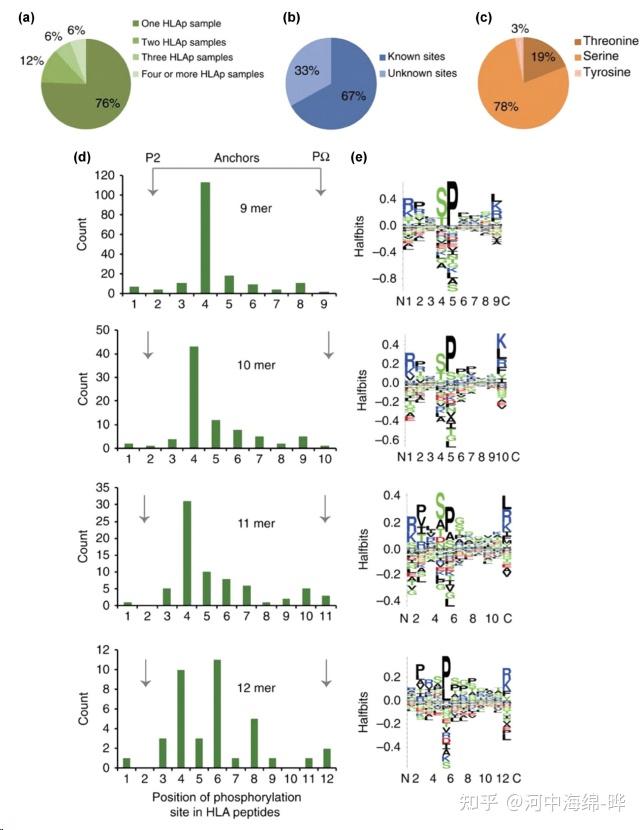
3.翻译后修饰(Post-Translational Modifications, PTMs)分析:质谱可用于检测和定位蛋白质的翻译后修饰,如磷酸化、乙酰化、泛素化等。通过对修饰位点进行定量分析,可以研究蛋白质的功能和调控机制。
4.蛋白质相互作用分析:质谱可用于研究蛋白质之间的相互作用。例如,可以通过免疫共沉淀(Immunoprecipitation, IP)或亲和纯化(Affinity Purification)等方法富集蛋白质复合物,然后用质谱进行蛋白质鉴定。这有助于揭示蛋白质之间的网络和通路。
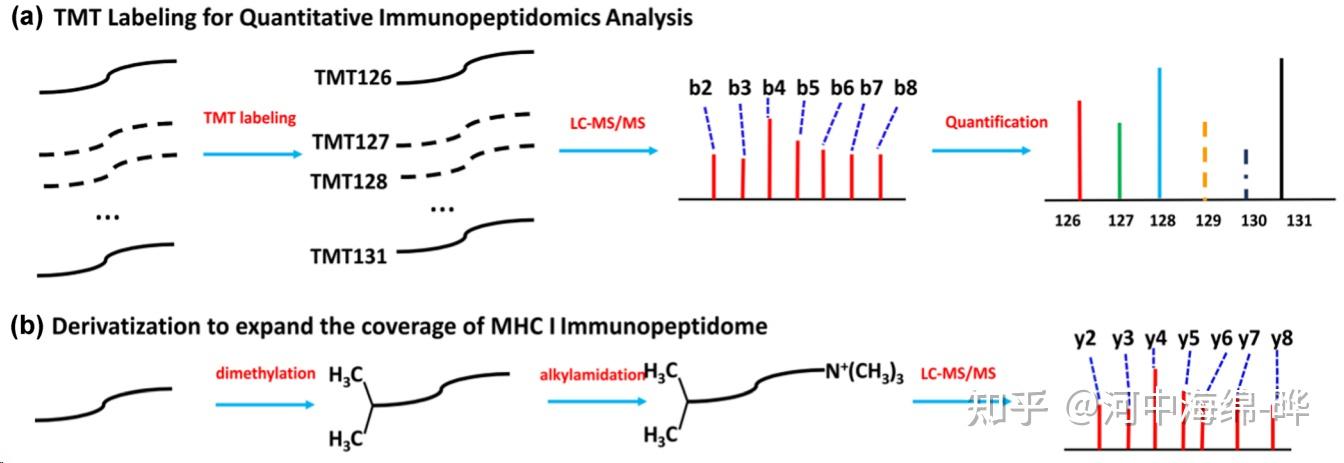
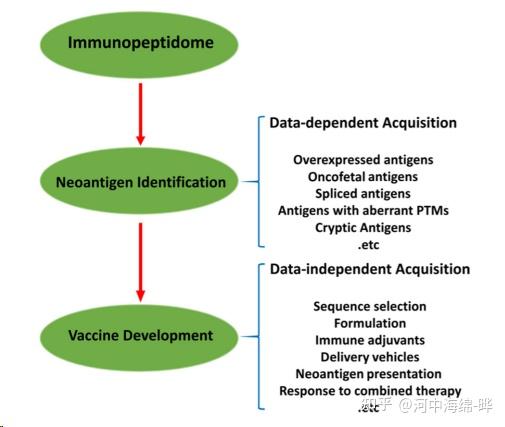
5.定量蛋白质组学:质谱可用于定量研究蛋白质的表达水平。通过标记(如同位素标记,如iTRAQ、TMT等)或标记自由(如标签自由定量,LFQ)方法进行蛋白质定量分析,可用于研究生物学过程、疾病状态和药物干预等条件下的蛋白质表达变化。定量蛋白质组学可以帮助了解生物学现象、病理过程以及药物作用机制。
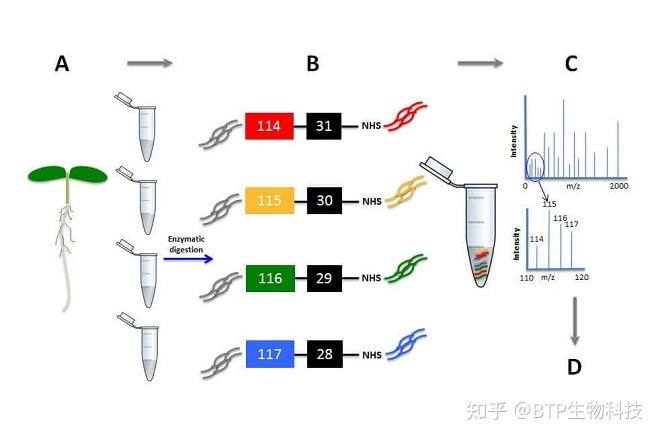
6.结构生物学:质谱在结构生物学中也有广泛应用,如氢/氘交换质谱(Hydrogen/Deuterium Exchange Mass Spectrometry, H/DX-MS)用于研究蛋白质的结构和动态性。此外,质谱也可用于研究蛋白质折叠、构象变化以及与其他生物大分子的相互作用。
7.顶点定向蛋白质酶切(Top-Down Proteomics):顶点定向蛋白质酶切是一种不需要对蛋白质进行酶切的质谱方法,直接对整个蛋白质分子进行测量。这种方法可以提供关于蛋白质的详细信息,包括翻译后修饰和剪切等。然而,由于其对质谱仪性能的高要求和数据处理的复杂性,目前顶点定向蛋白质酶切的应用相对较少。
8.蛋白质组学技术的发展:质谱技术不断发展,蛋白质组学研究也在不断推进。例如,新的数据独立采集(Data-Independent Acquisition, DIA)方法提高了质谱分析的准确性和可重复性。此外,质谱在研究蛋白质与小分子、药物筛选和药物靶点鉴定等领域也具有广泛应用。
相关技术文章分享:
蛋白质质谱鉴定
蛋白测序
定量蛋白组分析
更多科研干货,实验资讯,欢迎关注“百泰派克生物质谱”公众号 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-10-13 07:00
发表于 2024-10-13 07:00


 发表于 2024-10-13 07:00
发表于 2024-10-13 07:00