常规聚合酶链反应 (polymerase chain reaction, PCR) 是 20 世纪 80 年代中期发展起来的体外核酸扩增技术。PCR 是在体外模仿体内的 DNA 复制条件进行 DNA 大量快速复制的过程,具有特异、敏感、产率 高、快速、简便、重复性好、易自动化等突出优点;可以从一根毛发、一滴血,甚至一个细胞中于数小时 内将所要研究的目的基因或某一 DNA 片段扩增至十万乃至百万倍,获得足量的 DNA 供分析研究和检测鉴 定。PCR 的特点是每次 PCR 反应合成的产物都能和其模板一起作为下一次 DNA 合成的模板,使靶 DNA 片段呈指数形式增加。
最初建立 PCR 是为了扩增已知序列的靶基因。因为在 PCR 方法问世以前,要获得一个靶基因,必须 建立基因文库,然后从成千上万的菌落中通过 Southern blot 杂交筛选含有靶基因的克隆。这样既费时又费 钱,特别是在克隆真核生物基因时难度更大。自从建立了 PCR 方法以后,使克隆已知序列的基因变得非常 容易。为了适应分子生物学的快速发展,PCR 方法也得到了长足发展,现在 PCR 已应用到生命科学的各 个领域。
一、PCR 技术的基本原理与过程
(一)PCR 技术的基本原理
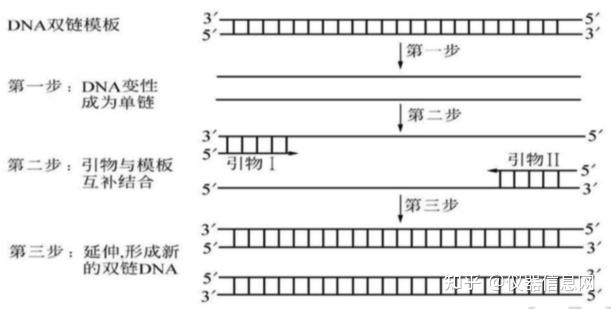
PCR 是一种选择性体外扩增 DNA 片段的方法,DNA 的合成过程与细胞内 DNA 的天然复制过程类似。 通过试管反应使极少量的基因组 DNA 样品中的特定基因片段,在短短几小时内扩增上百万倍。PCR 扩增 的特异性是由与靶序列两端区域互补的寡核苷酸引物决定的,即 PCR 中 DNA 的合成过程均以两个引物特 异结合处为起点;一旦合成产生了以两个引物为端点的特异的 DNA 分子,则这种分子在以后的热循环中 理论上将以几何指数方式增加,从而最终累积的 PCR 产物绝大多数是以两个引物为端点特异的 DNA 片段。 PCR 就是体外模仿体内的 DNA 快速复制过程,只不过 DNA 的变性、引物退火以及新 DNA 链的合成均采 用温度的升降来控制,同时通过反复的加热、冷却使反应体系的温度反复进行高温-低温-中温的热循环, 使 DNA 的复制过程不断重复进行;而且由于每次新合成的 DNA 片段都和其模板一样可以作为下一次热循 环过程中 DNA 复制的模板,因此可以实现对特定核苷酸片段的指数级扩增(图 4.1)。
图 4.1 PCR 原理示意
(二) PCR 反应过程
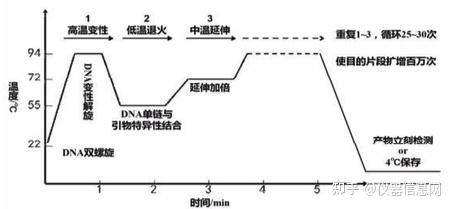
PCR 进程由变性-退火-延伸三个基本反应步骤反复重复进行;1高温变性(denaturation),PCR 反应液 中的模板 DNA 加热至 93~96°C一定时间(10~30s)后,模板 DNA 双链或经 PCR 扩增形成的双链 PCR 产物 发生双螺旋的氢键断裂而变性解链变成单链 DNA,成为合成 DNA 的活性模板。2低温退火(annealing), 在反应体系温度降至特定的温度 (一般为 50~55°C)时,经加热变性成单链的模板 DNA 与引物以碱基互补 配对的方式特异性结合,形成引物模板复合物。由于引物浓度远大于模板浓度,该过程是由引物驱动的。 3中温延伸(elongation),在反应体系升温到60~75°C时单链DNA模板-引物结合物可被 DNA聚合酶识别, 并在 Taq DNA 聚合酶的作用下,以四种脱氧核糖核苷三磷酸(dNTP)为反应原料,单链 DNA 为模板,按碱 基互补配对方式,从 5'向 3'方向合成一条新的与模板 DNA 链互补的 DNA 链,使单链 DNA 模板重新成为 双链,从而完成 DNA 靶序列的半保留复制。PCR 的变性-退火-延伸三个反应步骤反复进行,使特异 DNA 片段拷贝数呈指数上升。这样经过 2~3h 可完成 30~40 个热循环,PCR 产物量最高可达起始模板量的 230 倍以上。
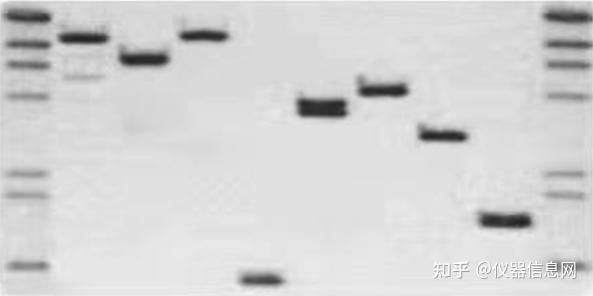
凝胶电泳(gel electrophoresis)是以凝胶为支撑物的区带电泳,常用的有琼脂糖凝胶和聚丙烯酰胺凝胶电 泳等。凝胶电泳是检测 PCR 产物最常用、最简便的方法。通过凝胶电泳检测,我们可以判断 PCR 反应扩 增产物的有无及大小。根据检测的目的基因不同,PCR 扩增后可产生长度不同的 DNA 扩增片段,其核苷 酸片段的分子量也有所不同,在凝胶电泳中分子量小的 DNA 片段移动速度快,分子量大的 DNA 片段移动 速度慢。因此,通过凝胶电泳判断 DNA 扩增片段的大小,可以满足检测的需要。常用的凝胶电泳有琼脂 糖凝胶电泳和聚丙烯酰胺凝胶电泳,前者主要用于 DNA 片段大于 500bp 者,后者主要用来检测小片段 DNA。
(一)琼脂糖凝胶电泳的原理与方法
1. 琼脂糖凝胶电泳原理
琼脂糖凝胶电泳(agarose gel electrophoresis )是一种简便、快速、常用的鉴定、 分离和纯化核酸的方法。对于分子量较大的样品,如大分子核酸、病毒等,一般可采用孔径较大的琼脂糖 凝胶进行电泳分离。琼脂糖凝胶电泳的分析原理与其他支持物电泳最主要的区别是:它兼有“分子筛”和“电 泳”的双重作用。
琼脂糖凝胶电泳的原理:琼脂糖凝胶电泳是以琼脂糖为介质,对不同大小的 DNA 或 RNA 实现分离的 一种电泳方法。琼脂糖是一种多糖,具有亲水性,不带电荷。使得 DNA 在碱性条件下使其在带负电荷(缓 冲液的 pH8.0)在电流作用下,以琼脂糖凝胶为介质,由负极向正极移动。根据不同的 DNA 分子片段的大 小和形状不同,在电场中泳动的速率也不相同,同时在样品中加入染料(如 EB 或花青素类染料)能够和 DNA 分子间形成络合物,经过紫外照射,可以看到 DNA 的位置(比对 marker 可知分子量大小),从而达到分离、 鉴定的目的。
2.琼脂糖凝胶电泳的操作步骤
琼脂糖凝胶电泳仪的组成有,电泳仪和电泳槽两个部分,配件有与电泳 槽配套使用的梳子,梳子的作用是在琼脂糖凝胶上形成点样孔,电泳仪组成见图 4.3。
图 4.3 电泳仪组成
操作步骤如下:
(1)材料准备:PCR 产物、电泳缓冲液、DNA 分子量 marker (用于指示 PC 产物的分子量大小)、溴 酚蓝(用于指示电泳的进度)。
(2)制胶
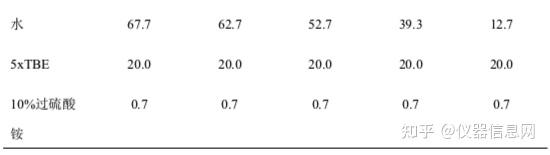
琼脂糖凝胶厚度约为 3~5mm。过薄则加样孔样品会溢出来,过厚观察时荧光穿透不强以至有些小片段 DNA 带型不易分辨。如配制 2%凝胶 100ml,称取琼脂糖 2g 于三角瓶中,加入 100ml 0.5×TBE 液,微波炉 加热 2-5min,使琼脂糖完全溶解(注意不要暴沸),置室温,等温度下降至 60°C时,加入终浓度 0.5μg/ml 的 EB 液,充分混匀后倒板(注意排除气体)。
(3)加样和电泳
上样时一般取 PCR 反应液 5~10μl,加入 3μl 溴酚兰液,充分混匀,加入凝胶加样孔中。电泳仪可用一 般稳压可调中压电泳仪,电泳工作液为 0.5×TBE 液,接通电源,使样品由负极向正极移动。60-100V 恒压 电泳约 30-60 分钟。
(4)结果观察
电泳完成后,还需要经过特殊的染色才能将 DNA 条带显示出来。琼脂糖凝胶电泳结果常用的显色方法是荧光染料染色法,实验室一般采用溴化乙锭、Gold View 或 Gel Red 荧光染料染色。 溴化乙锭(Ethidium Bromide,EB)是一种荧光染料,可嵌入核酸双链的碱基对之间,在紫外线激发下,发出红色荧光,从而使肉眼无法直接观察到的 DNA 可以在紫外光照射下被观察到。琼脂糖凝胶 EB 染色 的方法可在凝胶电泳液中加入终浓度为 0.5ug/ml 的 EB,有时亦可在电泳完成后,将凝胶浸入该浓度的溶 液中染色 10~15min,于紫外透射仪上观察照相。PCR 产物经 EB 染色,紫外透射仪下观察,初步判断产物 的特异性。PCR 产物片段的大小应与预计的一致,特别是多重 PCR,应用多对引物,其产物片段都应符合 预期的大小。溴化乙锭(EB)是一种强致癌剂,长期操作 EB 或者所处实验环境中有暴露的 EB,都会对 操作人员造成潜移默化的危害,尤其是遗传方面。EB 的废液处理不当,也对周围存在生物安全隐患,污染环境。
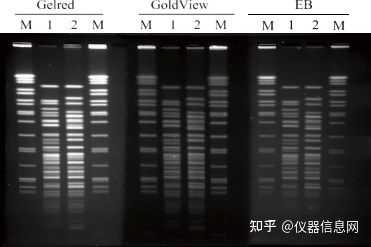
新的核酸染料 Gel Red 可以取代 EB, Gel Red 无论用于预制凝胶染色还是凝胶电泳后染色,都表现出 了极高的灵敏度。有报道 Gold View 的毒性低于 EB,但仍然具有毒性。而 Gel Red 的特殊化学结构使其难 以穿透细胞膜进入细胞,从而降低了染料的细胞毒性,更加安全。见图 4.4。
图 4.4 Gel Red、GoldView 和 EB 三种染料 PFGE 染色结果对比
(二) 聚丙烯酰胺凝胶电泳的原理与方法
1.聚丙烯酰胺凝胶电泳原理
聚丙烯酰胺凝胶电泳的原理与琼脂糖凝胶电泳类似,只不过聚丙烯酰胺 凝胶是由丙烯酰胺和甲叉双丙烯酰胺聚合而成的立体网状凝胶,其对 DNA 的有效分离范围和分辨率与琼 脂糖凝胶电泳有所不同。与琼脂糖凝胶电泳相比,聚丙烯酰胺电泳具有如下的优点:1分辨率很高,长度 仅相差 0.2%(即 500bp 中的 1bp)的 DNA 分子即可分开;2能装载的 DNA 量远远大于琼脂糖凝胶,多达 10ug 的 DNA 可以加样于聚丙烯酰胺凝胶的一个标准样品槽(1cmx1cm)而不致显著影响分辨力;3从凝胶 中回收的 DNA 纯度很高。
凝胶电泳检测电泳后的扩增产物显示的方法主要有溴化乙锭(EB)染色和银染法两种。丙烯酰胺有神经毒作用,可经皮肤吸收,并可在体内蓄积。配制和使用丙烯酰胺及亚甲双丙烯酰胺时必须戴手套和面具。 取用含上述化学药品的溶液也要戴手套。尽管可以认为聚丙烯酰胺无毒,但鉴于其中可能含有少量未聚合 的丙烯酰胺,故仍应小心处理。
2.聚丙烯酰胺凝胶电泳的操作步骤
(1)清洗
用蒸馏水将胶膜、电泳槽和梳子冲洗干净,装好胶膜,放在水平桌面上。
(2) 制胶
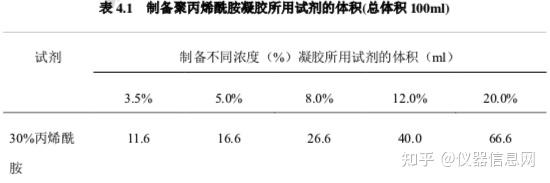
根据核酸分子质量的大小选择配置不同浓度的丙烯酰胺凝胶,一般 200-400bp 的 DNA 片段可配制 6%—8%,根据表 4.1 计算各组分的用量;在干净的三角烧瓶中分别加入上述各组分,充分混匀,小心倒 入胶膜中,插上梳子,待凝固。
(3)加样和电泳
向电泳槽中倒入 1×TBE,其量以没过胶面 2mm 为宜,小心移去梳子(如样品孔内有气泡应除去);在 DNA 样品中加入 0.2 体积的载样缓冲液,混匀后,加入样品孔内;接通电源,一般红色为正极、黑色为负 极,切记 DNA 样品由负极往正极泳动(靠近加样孔的一端为负),电压为 1-5V/cm(长度以两个电级之间 的距离计算);根据指示剂泳动的位置,判断是否终止电泳,一般 200-400bp 的 PCR 产物 100V 电压,电 泳 20-40min。
(4)结果观察
电泳结束后切断电源,取出胶膜,浸入含 0.5ug/ml 的溴化乙锭溶液中染色 10~20min;紫外透射仪上观察电泳带及其位置并与核酸分子质量标准比较被扩增产物的大小。
二、核酸探针杂交检测
(一)核酸探针杂交检测法
核酸探针杂交检测法是鉴定 PCR 扩增产物特异性的一种重要手段,常用的方法有:
1. 点杂交
点杂交是核酸杂交中比较简单的一种技术,本质上也是单链核酸分子通过碱基互补而形成 双链核酸的过程,当某单链分子被标记后,就可以检测与其互补的分子。由于杂交分子预先没有进行像 Southern 杂交和 Northern 杂交之前的电泳分离,也没有像原位杂交那样,杂交一方的分子已经表现出组织 细胞分布的位置特异性,所以点杂交的结果判断完全取决于杂交信号的强弱,其信息量没有其它杂交形式 的信息量丰富。但点杂交的操作比较简单、结果直观、适用于大批样品的检测,因此它在许多方面也得到 了广泛的应用。
2. 反向斑点杂交
利用生物素等标记的探针和特异性扩增 PCR 产物(靶 DNA 序列)杂交,但是不同 于一般的斑点杂交。一般的点印迹杂交是靶 DNA 固定于硝酸纤维素膜或尼龙膜上,标记的探针和靶 DNA 杂交显色。这种方法每次杂交反应只能判断待测 DNA 是否含有某一种探针的同源序列,对于某些基因座 位可能含有十几至几十个等位基因,用点杂交的方法就会很繁杂,甚至可能做不到。而反向斑点杂交解决 了这一难题。反向斑点杂交是先将待用的探针分别点到硝酸纤维素膜或尼龙膜上,每个探针一个点并编上 号,再将待测的 DNA 样本与之杂交,这样待测样本就会与具有同源序列的探针结合,经洗涤去除未结合 的 DNA 样本,由于待测的 DNA 样本具有生物素类的标记物,再经相应的显色反应就能显出杂交信号。这 样一次就可以判断某一基因座位生物大部分或全部等位基因。因此,可以用来鉴定扩增产物的特异性,有助 于产物的分型及法医学鉴定,且可以同时检测多个突变位点或多种病原体。
3. Southern 杂交
Southern 印迹杂交是进行基因组 DNA 特定序列定位的通用方法。一般利用琼脂糖 凝胶电泳分离经限制性内切酶消化的 DNA 片段,将胶上的 DNA 变性并在原位将单链 DNA 片段转移至尼 龙膜或其他固相支持物上,经干烤或者紫外线照射固定,再与相对应结构的标记探针进行杂交,用放射自 显影或酶反应显色,从而检测特定 DNA 分子的含量。
图 4.5 Southern 印迹杂交显色图片
Southern 印迹杂交技术的基本原理是:具有一定同源性的两条核酸单链在一定的条件下,可按碱基互 补的原则形成双链,此杂交过程是高度特异的。由于核酸分子的高度特异性及检测方法的灵敏性,综合凝 胶电泳和核酸内切限制酶分析的结果,便可绘制出 DNA 分子的限制图谱。但为了进一步构建出 DNA 分子 的遗传图,或进行目的基因序列的测定以满足基因克隆的特殊要求,还必须掌握 DNA 分子中基因编码区 的大小和位置。有关这类数据资料可应用 Southern 印迹杂交技术获得。Southern 印迹杂交技术包括两个主 要过程:合到一定的固相支持物(硝酸纤维素膜或尼龙膜)上,即印迹;二是固定于膜上的核酸与同位素 标记的探针在一定的温度和离子强度下退火,即分子杂交过程。
(二) 核酸探针的标记
核酸探针的标记可分为放射性标记和非放射性标记两类。
1.放射性标记
常见的放射性核素标记物有 32P、3H 和 35S。放射性核素标记的探针具有高度的特异性 和敏感性,可以检出 1~10 ug/uL 的高等生物基因组 DNA 中的单拷贝基因,然而由于放射性对环境污染限 制了其应用。
2.非放射性标记
放射性标记核酸探针在使用中的限制,促使非放射性标记核酸探针的研制迅速发展, 在许多方面已代替放射性标记,推动分子杂交技术的广泛应用。目前已形成两大类非放射标记核酸技术, 即酶促反应标记法和化学修饰标记法。
酶促反应标记探针是用缺口平移法,随机引物法或末端加尾法等把修饰的核苷酸如生物素-11-dUTP 掺 入到探针 DNA 中,制成标记探针,敏感度高于化学修饰法,但操作程序复杂,产量低,成本高。化学修 饰法是将不同标记物用化学方法连接到 DNA 分子上,方法简单、成本低,适用于大量制备(>50μg)如光 敏生物素标记核酸方法,不需昂贵的酶,只在光照 10~20min,生物素就结合在 DNA 或 RNA 分子上。
非放射性标记核酸探针方法很多,现介绍常用的几种方法如下:
(1) 生物素标记核酸探针方法:生物素标记的核苷酸是最广泛使用的一种,如生物素-11-dUTP,可用 缺口平移或末端加尾标记法。实验发现生物素可共价连接在嘧啶环的 5 位上,合成 TTP 或 UTP 的类似物。 在离体条件下,这种生物素化 dUTP 可作为大肠杆菌多聚酶 I(DNA 酶 I)的底物掺入带有缺口的 DNA 或 RNA,得到生物素标记的核酸探针。这种标记方法称为缺口平移法。用标记在 DNA 上的生物素与链霉亲 合素-酶(过氧化物酶或碱性磷酸酶)标记物进行检测。
(2) 光敏生物素标记核酸探针:光敏生物素有一个连接臂,一端连接生物素,另一端有芳基叠氮化合 物。在可见光照射下,芳基叠氮化合物可能变成活化芳基硝基苯,很易与 DNA 或 RNA 的腺嘌呤 N-7 位置 特异结合,大约每 50 个碱基结合一个生物素,所以只用于标记大于 200 个核苷酸的片段。光敏生物素的 醋酸盐很易溶于水,与核酸形成的共价结合很稳定。此法有以下优点:方法简便易行,快速省时,不需昂 贵的酶和 dUTP 等;只需光照,探针稳定,-20°C可保存 12 个月以上。适用于 DNA 和 RNA,抗体和酶等 的标记。在原位分子杂交,斑点杂交和 Southern 印迹杂交中应用,其特异性和灵敏性较高,价廉易购。
(3) 生物素-补骨脂素标记法 生物素-补骨脂素是另一种生物素光敏物质,在长波长紫外线照射下与嘧 啶碱基发生光化学反应,加成到 DNA 中,去除小分子后,得到生物素标记核酸探针。此法可标记单链或 双链 DNA 或 RNA,及寡核苷酸。灵敏度与放射性探针相当。
(4) 生物素-α-氨基乙酸-N-羟基琥珀标记化学修饰的 DNA 法 此法是在亚硫酸盐催化下,生物素酰肼 可置换寡核苷酸探针中胞嘧啶上的氨基,使生物素结合到 DNA 分子上而制成生物素化 DNA 探针。此法优 点是采用通用试剂和技术,检出敏感。
第 3 节 PCR 扩增产物纯化
PCR 反应完成目标 DNA 的扩增后,PCR 产物可以用于进一步的研究,例如用于 DNA 测序、克隆、 分析基因的功能和表达等。然而,通过 PCR 反应后反应体系中仍然存在具有活性的 DNA 聚合酶,剩余 的 dNTP、引物以及反应缓冲体系中的各种金属离子等,因此我们必须通过一定的方法从反应体系中提纯 PCR 反应的扩增产物,才有利于进一步的分析研究。
一、产物直接纯化
PCR 扩增完成后需要进行琼脂糖凝胶电泳检测,在条带单一无其他杂带的情况下,可以直接对产物进 行纯化,常见的纯化方法有:
(一)硅胶层析柱法
原理:大多数离心柱中吸附 DNA 的是一层硅胶膜,实际上就是一层玻璃纤维,其表面有大量修饰的 硅羟基(Si-OH),硅羟基在溶液中解离后带负电,然后与带正电盐离子、带负电 DNA 形成电桥,从而吸 附 DNA,使得 DNA 双链变单链,不会被生物大分子溶剂洗脱,但能经水溶性缓冲液水化后,被定量回收, 从而实现纯化分离。
(二)磁珠法
原理:磁珠是有磁性能吸附 DNA 的物质,其内部核心是铁离子,在磁场作用下可以吸附在磁体上。 在核心外,经过羧化带有羧基基团,在特定的离子条件下,可以与 DNA 结合。结合后,在外磁场的作用 下,磁珠与 DNA 结合体移动到磁体周围,可以很方便去除其他多余的液体,不能与磁珠结合的所有杂质 都随水溶液一并去除。然后,在洗脱条件下,DNA 与磁珠分离,溶解在水中,将溶解有 DNA 的溶液转移, 就得到了纯化后的 DNA 样本。
(三)其他纯化方法
1. 渗透液结合醇沉纯化
原理:利用分子大小不同,小分子的物质能够通过渗透膜溶解到大量的溶液中去,而大分子的物质不 能通过渗透膜,所以继续留存在透析袋中,通过乙醇沉淀最后达到去除小分子物质,得到纯化的有机大分 子的效果。
2. 直接沉淀纯化
原理:DNA 分子在高盐离子醇溶液中会被沉淀出来,而其他物质继续溶解在溶液中,沉淀出来的 DNA 分子经过离心与溶液成分分开,达到纯化的目的。
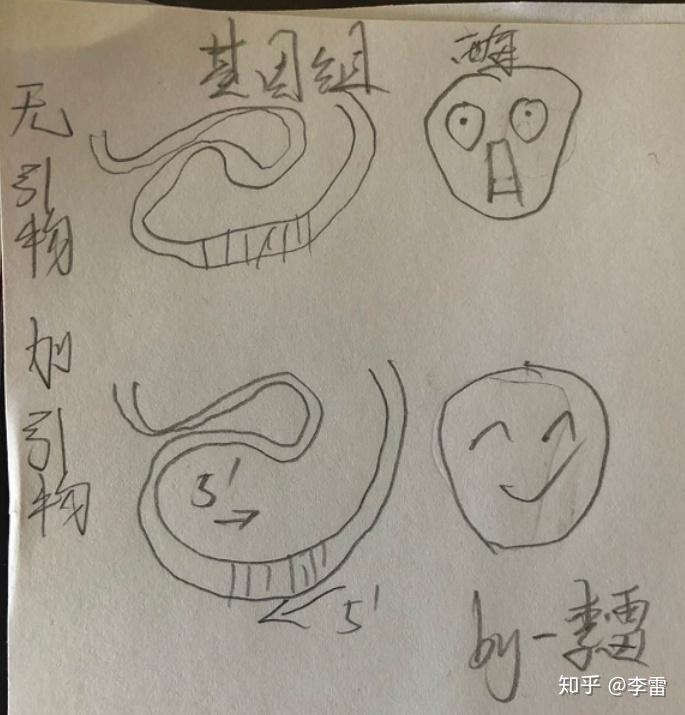
PCR即聚合酶链式反应,可以看做一种生物体外的特殊DNA复制。PCR的基本原理类似于DNA的天然复制过程。即DNA聚合酶结合到模板 DNA 后,可以将游离的脱氧核苷酸连接起来,形成与模板 DNA 互补的 DNA 单链。 但是DNA复制都只能沿着已存在的核苷酸链延伸,不能直接从头合成 DNA 链。
在体内进行DNA复制的时候,RNA聚合酶可以先与DNA模板结合,合成一小段 RNA 链,作为引物;紧接着 DNA 聚合酶再以RNA引物的3′端作为起始端,来合成新的DNA链;当DNA复制完成后,RNA引物将会被DNA聚合酶I或者核糖核酸酶切掉。
而在体外进行 DNA 复制时候,我们可以直接以人工合成的一段 DNA 片段作为引物,在复制结束时,就不需要再将引物切掉了。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-10-3 19:43
发表于 2024-10-3 19:43