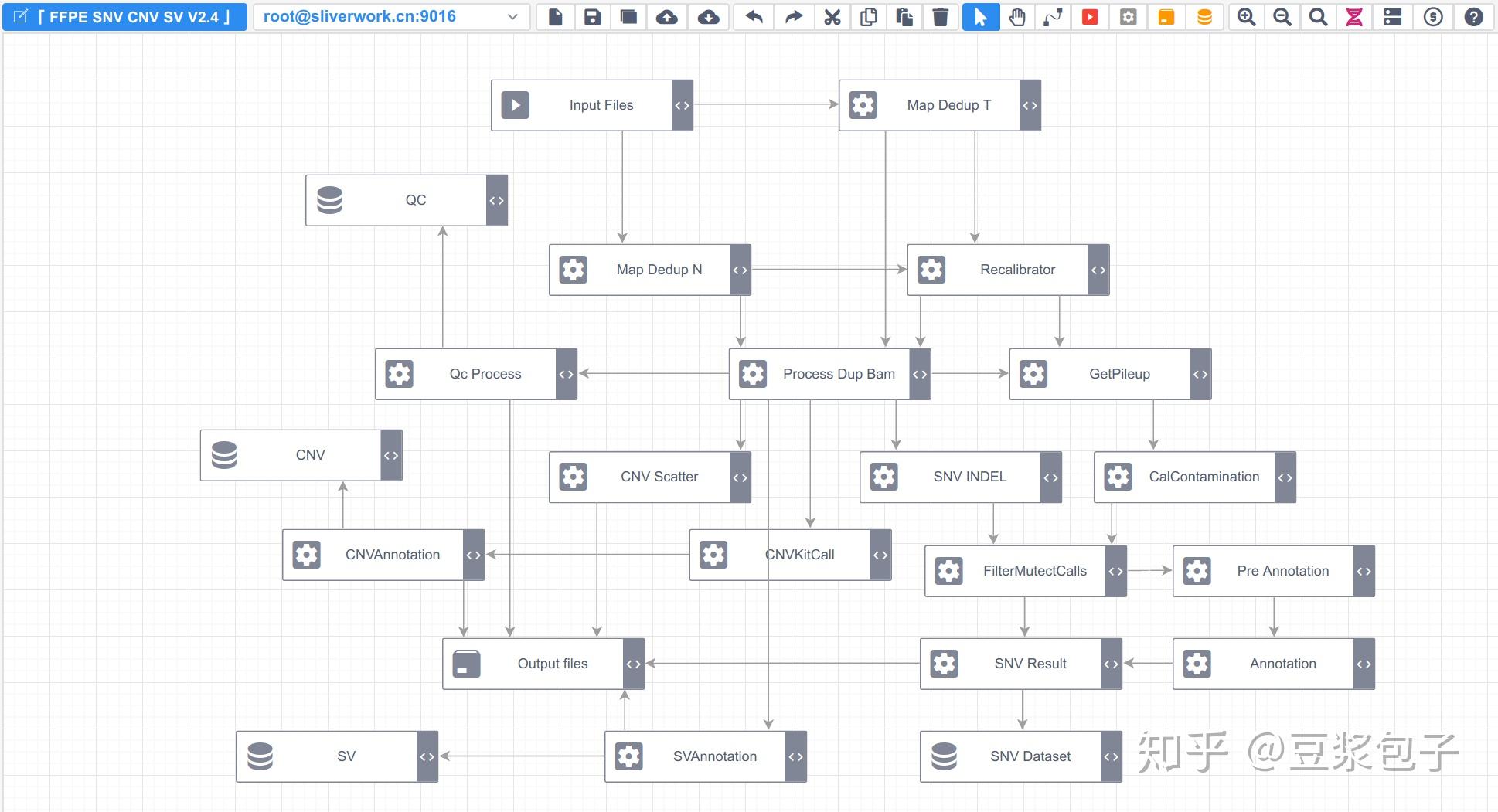
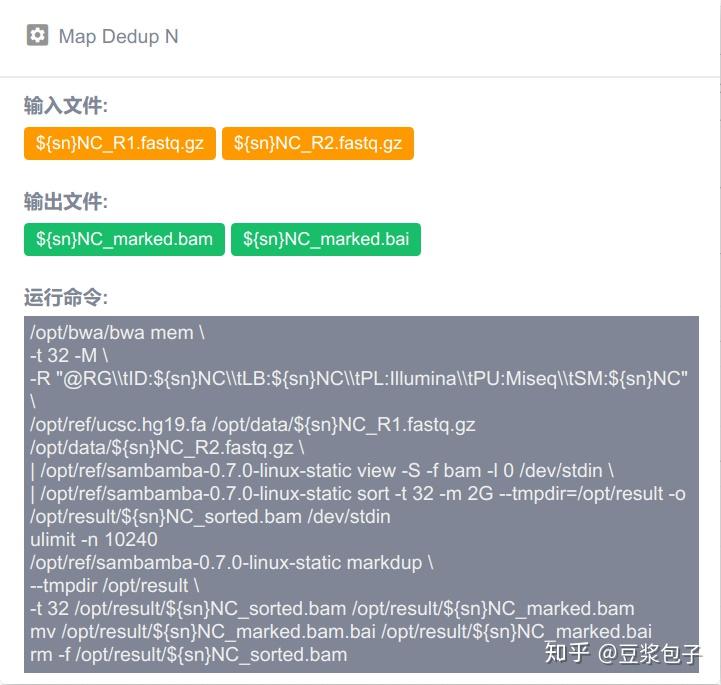
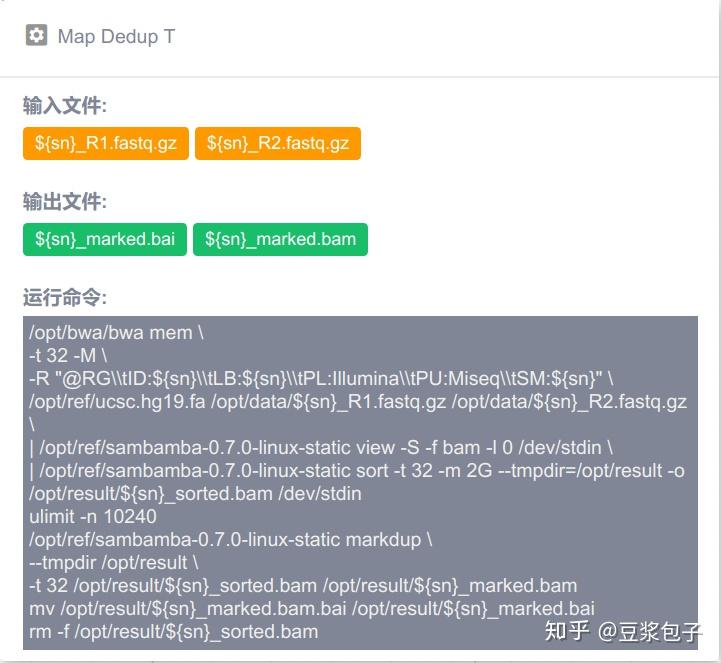
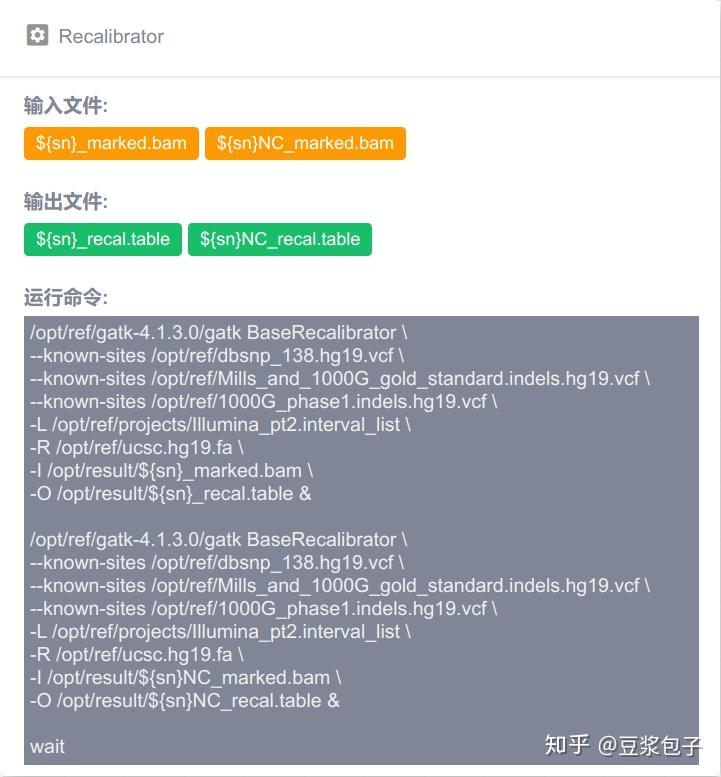
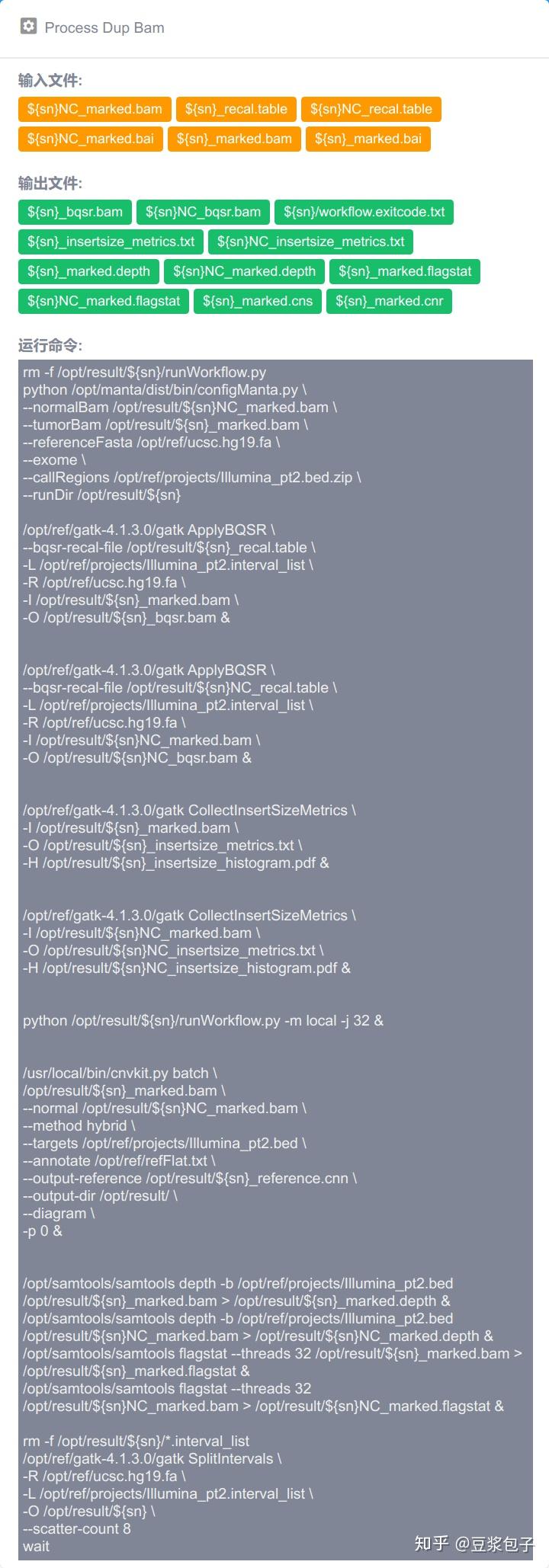
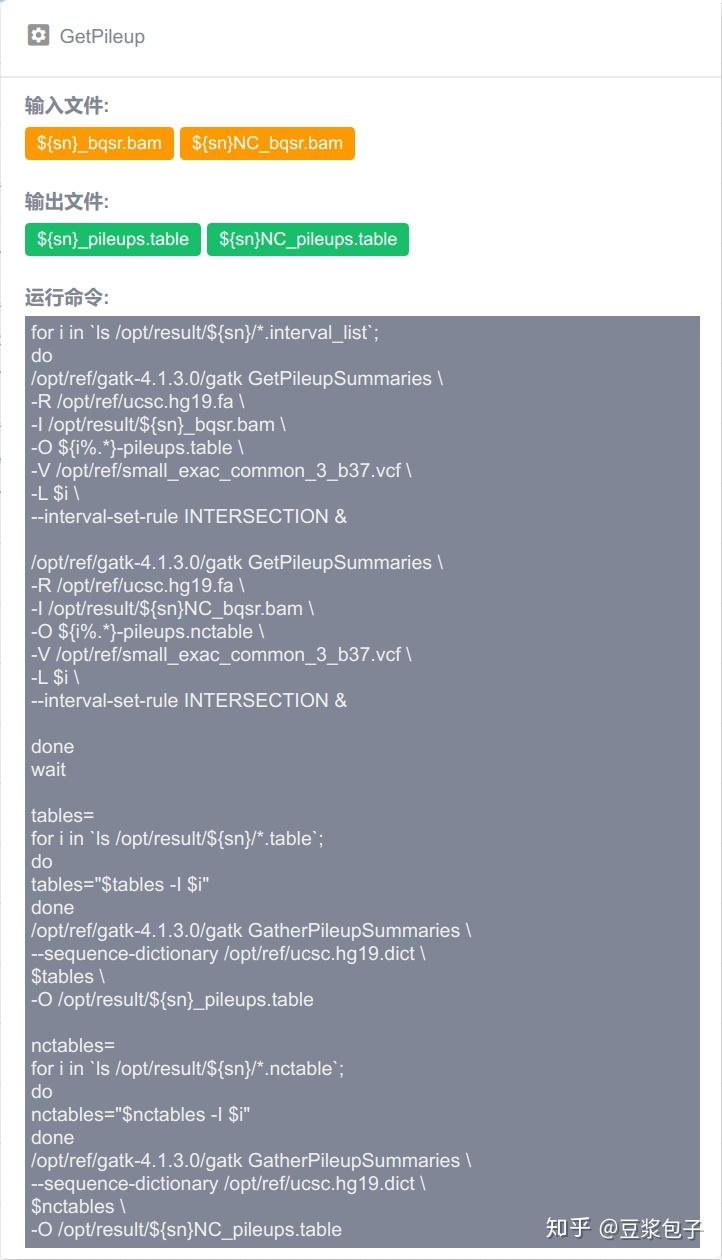
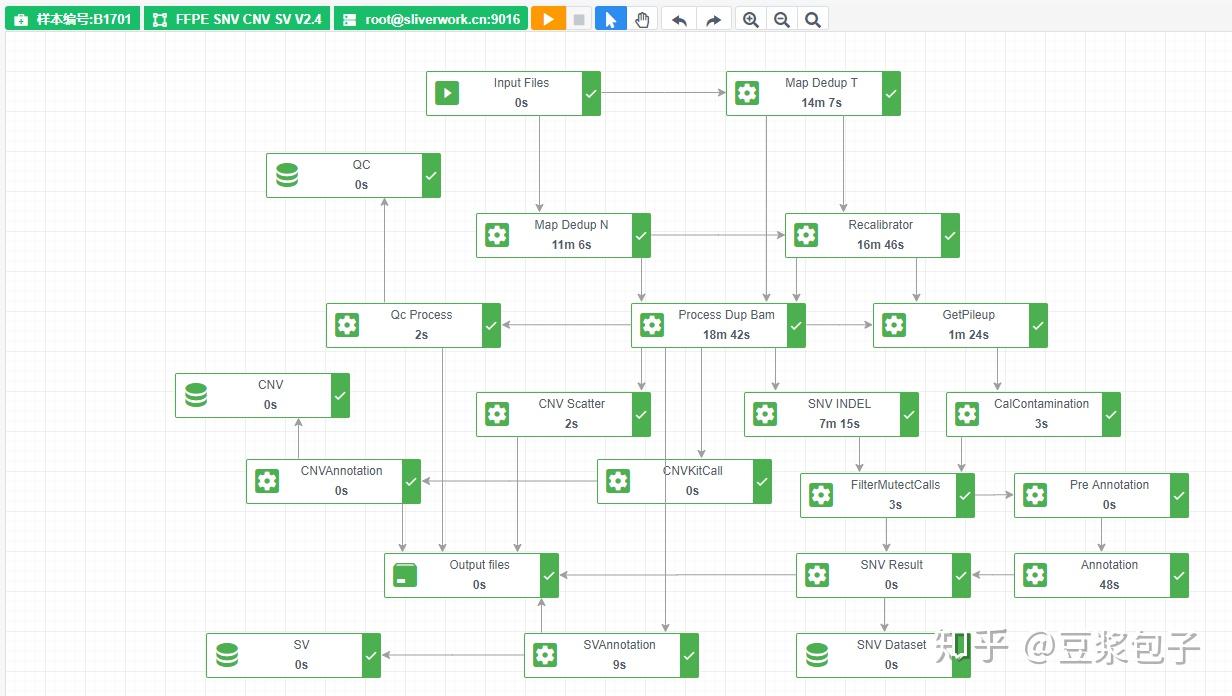
优化的方向:实际运行GATK4.X的工具如Mutect2时,发现其运行效率相当低,从CPU占用率,内存占用,硬盘I/O都占用很低,起初自己DIY时候,将要分析的bed/interval_list文件按照染色体编号拆分(不太确定分析结果的一致性,所以比较谨慎),然后并行分析,最后将结果合并。后来GATK从4.0.6.0升级到4.1.3.0时候发现官方的best practice pipeline也做了类似的处理,这里就有了优化的空间。
#生成合并参数,运行MergeMutectStats将状态文件合并
rm -f ${result}/${sn}_bqsr.vcf.gz.stats
stats=
for z in `ls ${result}/${sn}/*_bqsr.vcf.gz.stats`;
do
stats="$stats -stats $z"
done
${tools.gatk} MergeMutectStats $stats -O ${result}/${sn}_bqsr.vcf.gz.stats

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-22 22:19
发表于 2024-9-22 22:19