登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
First-in-class 药物!以其首创性、独特机制和突破性疗效,引领医药创新前沿。本期汇总了 2024 上半年 FDA 获批的 First-in-Class 小分子,快来看看吧!
01
FIC 分子回顾
FDA 的 FIC (First-in-class) 分子指的是具有全新、独特作用机制的药物,这些药物是首个能够治疗某种疾病的药物。FIC 药物因其创新性,对于提供新的治疗机制和满足未被现有药物满足的医疗需求具有关键作用。
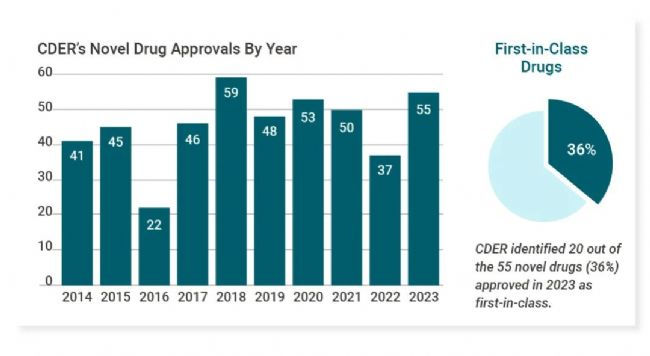
2023 年,FDA 共批准了 55 种新药,其中 20 款属于 FIC 药物,占比 36%,具体为 Trofinetide (Daybue),Taurolidine (Defencath),Iptacopan (Fabhalta),Sparsentan (Filspari),Filsuvez,Daprodustat (Jesduvroq),Leniolisib (Joenja),Lamzede,Perfluorohexyloctane (Miebo),Nirogacestat (Ogsiveo),Nirmatrelvir (Paxlovid),Tofersen (Qalsody),Rivfloza,Omaveloxolone (Skyclarys),Palovarotene (Sohonos),Talvey,Capivasertib (Truqap),Veopoz,Fezolinetant (Veozah),Lotilaner (Xdemvy)[1]。
图 1. FDA-CDER 近年新药批准情况 & 2023 年 FIC 品种占比。
2024 年上半年,FIC 分子继续在新药研究领域占据核心地位,具有潜力的 FIC 单靶点和双靶点分子被陆续报道,包括它们的作用机制、研发进展和潜在临床应用。
02
单靶点 FIC 产品介绍RORγ 共价抑制剂
核受体受体相关孤儿受体γ (RAR-related orphan receptor gamma, RORγ) 是一种配体依赖性转录因子,通过驱动雄激素受体 (Androgen receptor, AR) 过表达,已被确定为去势抵抗性前列腺癌 (Castration-resistant prostate cancer, CRPC) 中的关键参与者,代表了晚期前列腺癌的潜在治疗靶点。
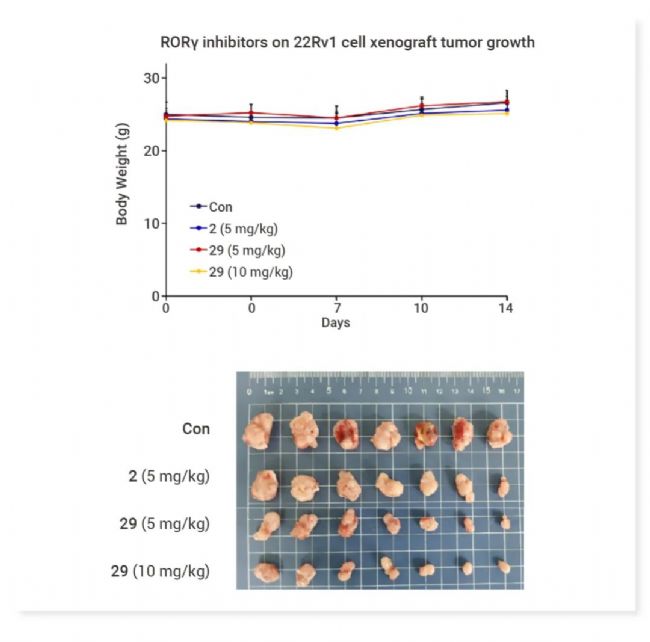
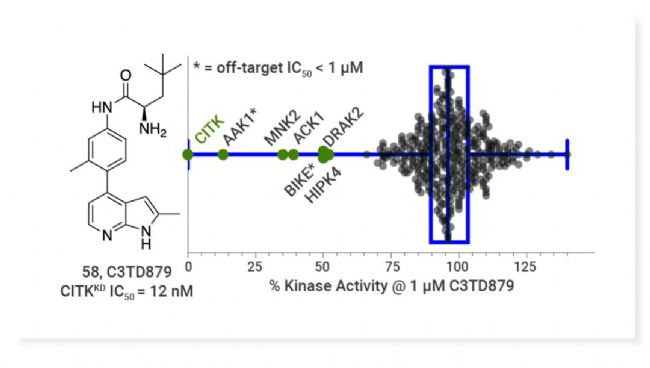
W6134 (29) 是 FIC RORγ 共价抑制剂,显著抑制 RORγ 转录活性并显著抑制 AR 和 AR 靶基因的表达水平。W6134 在抑制 CRPC 细胞系的增殖和集落形成以及诱导细胞凋亡方面表现出较阳性对照及非共价对照更优异的活性,且能显著抑制 22Rv1 小鼠肿瘤异种移植模型中的肿瘤生长,并具备良好的安全性[2]。图 2. W6134 (29) 显著抑制肿瘤体积且对小鼠体重无明显影响[2]。 CITK 抑制剂 Citron kinase (CITK) 是一种调节胞质分裂的 AGC 家族丝氨酸/苏氨酸激酶,在细胞的多种生物学过程中发挥作用,包括细胞骨架的重组、细胞运动、增殖和分化等。尽管敲除实验表明 CITK 是抗癌靶点,但其研究寄出往往集中于泛靶点抑制剂,尚不存在选择性 CITK 抑制剂。 Joshua 等以具有弱脱靶 CITK 活性的激酶抑制剂 Y-39983 为基础,成功开发出 FIC CITK 抑制剂 C3TD879[3]。C3TD879 是一种 I 型激酶抑制剂,可有效抑制 CITK 催化活性 (IC50= 12 nM),在细胞中直接与全长人类 CITK 结合 (NanoBRET Kd< 10 nM),并表现出良好的体内 DMPK 特性。 图 3. C3TD879, 一流的 CITK 抑制剂[3]。 ALK2 抑制剂 Activin receptor-like kinase 2 (ALK2) 是一种 I 型骨形态发生蛋白受体,在细胞中的作用主要涉及控制骨骼、心脏、大脑和其他组织发育的生物过程[4]。ALK2 通过响应配体的结合,传导成骨信号,参与调节细胞的增殖、分化和组织形成。ALK2/ACVR1 作为一种跨膜激酶受体,对于 TGF-β 家族成员的信号传导至关重要[5]。 Héctor 等对 M4K2009 进行新型支架修饰,得到了对 ALK2 效力更优异,且具有脑渗透性的 M4K2306,有作为 FIC 产品的潜力[6]。 GCN2 激酶激活剂 综合应激反应 (Integrated stress response, ISR) 是细胞应对各种应激条件的适应性机制。ISR 通过四种丝氨酸/苏氨酸激酶家族感应应激,其中之一为 General control nonderepressible 2 (GCN2) 激酶。GCN2 激酶响应氨基酸缺乏,可以磷酸化真核起始因子 2α (eIF2α),从而抑制全局蛋白质的翻译并促进特定应激相关基因的表达,包括转录因子 ATF4 (决定 ISR 和细胞命运的关键效应物)。 GCN2 激酶在免疫细胞中也扮演着多重角色。它参与调节巨噬细胞的功能极化和 CD4+ T 细胞亚群的分化。在肿瘤微环境中,GCN2 的激活能够导至 T 细胞无反应和细胞凋亡,增强骨髓来源抑制细胞 (MDSC) 的免疫抑制作用以及肿瘤细胞的存活。因此,GCN2 抑制可能具有直接的抗癌作用和免疫激活作用[7]。
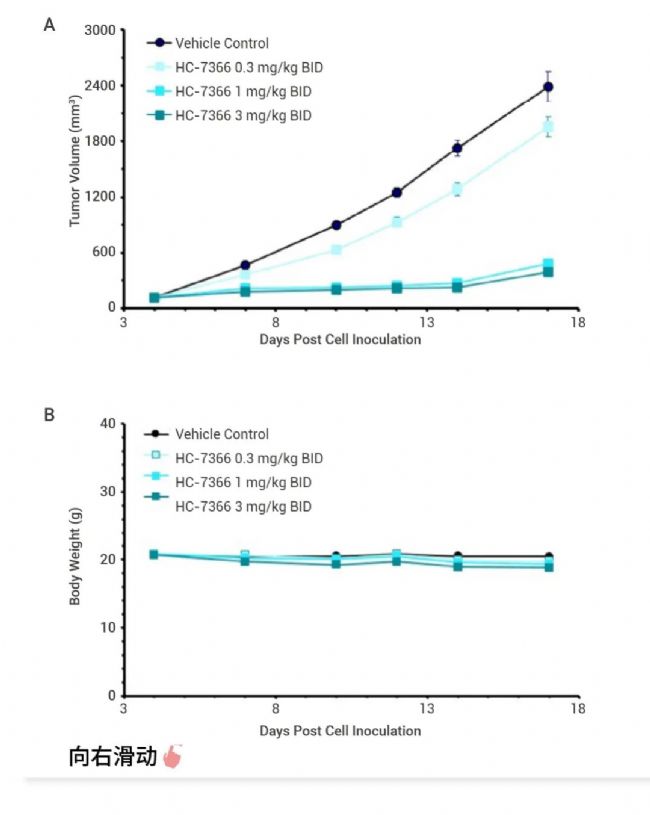
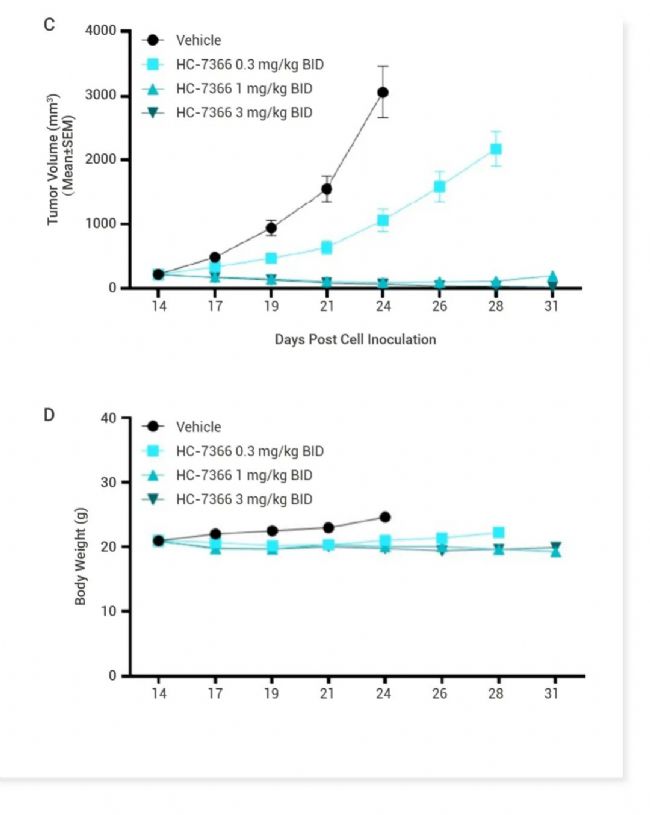
图 4. HC-7366 显著抑制肿瘤体积[8]。 A-B. HC-7366 在 HT1080 纤维肉瘤异种移植模型中的体内疗效。(A) 肿瘤体积与时间的关系; (B) 体重与时间的关系。C-D. HC-7366 在 MOLM-16 异种移植模型中的体内疗效。(C) 肿瘤体积与时间的关系;(D)体重与时间的关系。
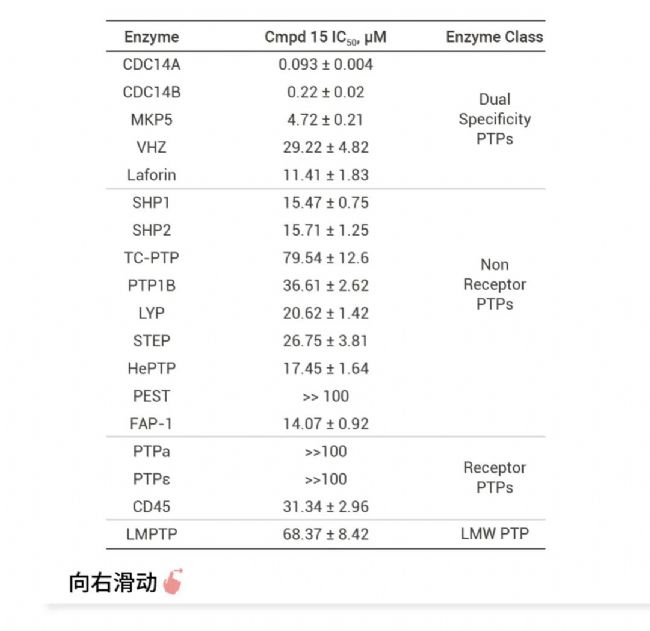
Thomson 等在已报道的 GCN2 配体的基础上进行多轮结构优化,得到 HC-7366 (29),一种口服生物可利用且有效的 GCN2 激酶激活剂[8]。单一药物 HC-7366 已证明具有强大的抗肿瘤活性,可在几种临床前肿瘤模型中导至肿瘤消退和完全缓解,已作为抗肿瘤疗法进入临床。 Cdc14 抑制剂 Cyclin-dependent kinase-counteracting (Cdc14) 是一种高度保守的丝氨酸/苏氨酸蛋白磷酸酶家族成员,在细胞周期调控中发挥重要作用。它可以通过降低 CDK 的活性来调节细胞周期,是真核细胞中重要的细胞周期调节蛋白。 同时,CDC14 在 DNA 损伤修复中也扮演着重要角色。CDC14 酶能够激活霍利迪连结体游离酶 (Holliday junction resolvase) Yen1,确保在细胞分裂前 DNA 断裂能够被完全修复,有助于预防染色体或染色体片段的缺失,这些错误与癌症和其他疾病有直接关系。这些发现表明,CDC14 不仅在细胞周期调控中发挥作用,还可能成为癌症治疗的潜在靶点,通过抑制 CDC14 的活性可能有助于阻止癌细胞的 DNA 修复,从而提高化疗的有效性[9]。 Dong 等通过基于片段和结构的设计策略,优化改进得到 CDC14A/B-IN-1 (Compound 15),一种 FIC,强效,口服生物利用度的人类 CDC14A/B 磷酸酶抑制剂,并证实了基于片段的设计策略在发现与开发具高活性、高选择性、良好的生物利用度的 PTP 抑制剂的适用性[10]。
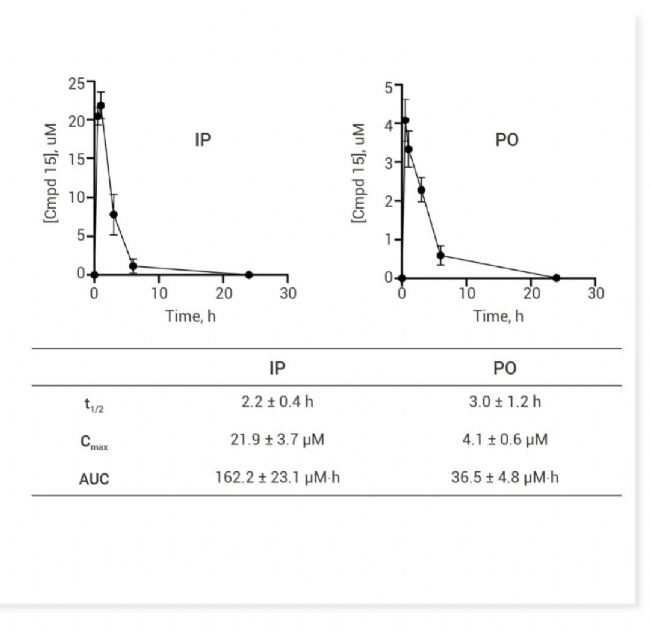
图 5. CDC14A/B-IN-1 (Cmpd 15) 对 16 种哺乳动物 ptp 的选择性及体内药代动力学数据[10]。
03
双靶点 FIC 产品介绍 EZH2/HSP90 双效抑制剂 增强子同源物 2 (EZH2) 是一种组蛋白甲基转移酶,主要通过多梳抑制复合体 2 (PRC2) 依赖的组蛋白 H3 中 27 位的赖氨酸三甲基化 (H3K27me3) 调控基因表达,参与了肿瘤的发生与发展[11]。 HSP90 在癌症治疗中是一个吸引人的靶点。抑制 HSP90 的表达可以影响多种致癌途径。然而,HSP90 抑制剂在临床开发中面临一些挑战,包括疗效低、毒性或耐药性问题。为了克服这些挑战,研究人员正在探索 HSP90 的双靶点抑制剂,以提高疗效和降低耐药性。例如,HSP90 与组蛋白去乙酰化酶 (HDAC) 抑制剂、微管蛋白抑制剂、拓扑异构酶 II (Topo II) 抑制剂等联合使用时,具有协同抗肿瘤作用。开发 HSP90 双重抑制剂被认为是一种有效的肿瘤治疗策略,可以增强疗效,同时降低耐药性[12]。
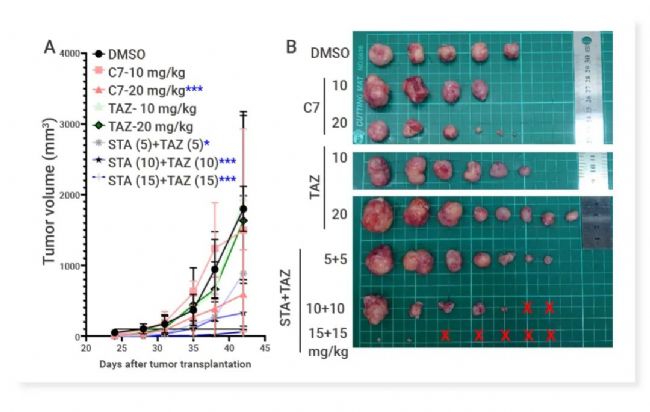
图 6. EZH2/HSP90-IN-29 (C7) 显著降低了 Pt#3-R 诱导的肿瘤生长[13]。
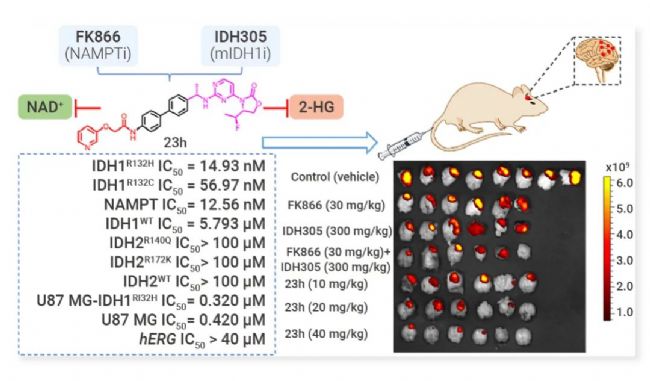
Sharma 等通过对 FDA 批准的 EZH2 抑制剂 Tazemetostat 和第 2 代 HSP90 抑制剂药效团片段进行结构分析,成功发现 FIC EZH2/HSP90 双靶点抑制剂 EZH2/HSP90-IN-29 (C7)[13]。EZH2/HSP90-IN-29 对 TMZ 耐药性胶质母细胞瘤 (GBM) 细胞系具有显著的细胞生长抑制作用,在移植了 TMZ 抗性 Pt3R 细胞的实验小鼠中表现出显著的体内抗 GBM 功效,具有作为抗 GBM 药物的潜力。 GPX4/CDK 双重抑制剂 GPX 4,谷胱甘肽过氧化物酶 4 (Glutathione peroxidase 4),是一种重要的抗氧化酶,利用谷胱甘肽(GSH)作为还原剂来清除脂质过氧化物,维持细胞内的氧化还原平衡。GPX4 是铁死亡的关键抵抗因子,通过催化脂质过氧化物的还原反应来抑制铁死亡的发生,因此被认为是癌症治疗的一个有前景的靶点[14]。 CDK,细胞周期依赖性激酶 (Cyclin-Dependent Kinases) 是一组在细胞周期调控中发挥关键作用的蛋白激酶,其异常活化与多种肿瘤的发生发展密切相关。 Zhu 等基于 ML162 (GPX4 抑制剂) 和靛玉红肟 (IO, CDK 抑制剂) 的重点结构片段,拼合并筛选出 FIC GPX4/CDK-IN-1 (B9)。化合物 B9 对四种细胞系均表现出最高的潜在细胞毒活性,并表现出对 GPX4 的优异抑制活性 (IC50= 542.5 nM) 和对 CDK 4/6 的选择性抑制活性 (IC50= 191.2, 68.1 nM)。B9 在 MDA-MB-231 细胞和 HCT-116 细胞中均能同时诱导铁死亡并将细胞阻滞于 G1 期。与 ML162 和 IO 相比,B9 在体内表现出更强的癌细胞生长抑制作用[15]。 IDH 1/NAMPT 双重抑制剂 异柠檬酸脱氢酶 1 (Isocitrate dehydrogenase 1, IDH1)是一种在细胞代谢过程中扮演重要角色的酶,参与三羧酸循环,负责催化异柠檬酸转化为 α-酮戊二酸 (α-KG) 的反应。Mutant IDH1 会将 α-KG 转化为致癌性代谢产物 2-羟基戊二酸 (2-HG),能抑制依赖于 α-KG 的双加氧酶,如组蛋白去甲基化酶和 DNA 去甲基化酶 TET 蛋白,导至 DNA 和组蛋白的高甲基化状态,从而影响细胞的分化和增殖[16]。 烟酰胺磷酸核糖基转移酶 (NAMPT) 是烟酰胺腺嘌呤二核苷酸 (NAD+) 挽救途径中的关键限速酶,NAMPT 通过催化合成 NAD+,参与调控细胞内多种生物学过程,包括 DNA 修复、细胞程序性死亡、代谢重编程等,这些过程对肿瘤细胞的代谢和增殖至关重要[17]。 Fei 等发现 FIC 小分子 Mutant IDH1/NAMPT-IN-1 (Compound 23 h) 对两个靶点表现出优异且平衡的抑制活性 (IC50分别为 14.93 nM, 12.56 nM),从而显著抑制 IDH1 突变的胶质瘤细胞 (U87 MG-IDH1 R132H) 增殖[18]。重要的是,23 h 能够穿过血脑屏障 (B/P 比为 0.76),并在 U87 MG-IDH1 R132H 原位移植小鼠模型中表现出显著的体内抗肿瘤效果 (20 mg/kg),且无任何显著毒性。这项概念验证研究证实了发现同时靶向 mIDH1 和 NAMPT 的小分子的可行性,为治疗胶质瘤提供了有价值的线索。
图 7. Mutant IDH1/NAMPT-IN-1 (23 h) 显著抑制 IDH1/NAMPT 靶点活性并具有体内抗肿瘤活性[18]。 Polθ/PARP 双重抑制剂
DNA 聚合酶 θ (Polθ) 近来成为一种参与 DNA 损伤修复的新的有吸引力的合成致死靶点。单独灭活 Polθ 或与 PARP 抑制剂联合灭活已证明对具有同源重组 (HR) 缺陷 (例如 BRCA 基因变异) 的肿瘤具有巨大的治疗潜力。
研究发现一种 FIC 高效双重 Polθ/PARP 抑制剂 Polθ/PARP-IN-1 (25 d),对 Polθ 和 PARP1 均表现出低纳摩尔抑制活性,且对具有 53BP1 缺陷的 PARP 抑制剂抗性的 MDA-MB-436 细胞保持敏感性[19]。
总而言之,Polθ/PARP-IN-1 在治疗 HR 缺陷型肿瘤 (包括获得性 PARP 抑制剂耐药的肿瘤) 方面具有开发潜力。
PD-L1/EGFR 抑制剂
细胞程序性死亡配体 1 (PD-L1) 是肿瘤细胞和肿瘤微环境中的非转化细胞上过表达的蛋白,它与肿瘤细胞的免疫逃逸密切相关[20]。表皮生长因子受体 (EGFR) 是一种重要的肿瘤相关信号通路,已成为一个重要的药物靶点。
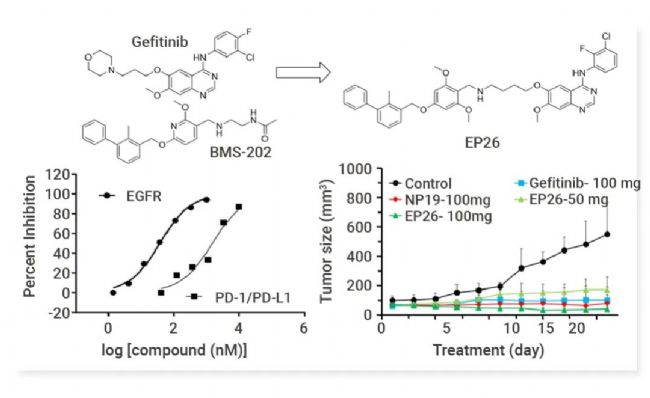
Yang 等基于 PD-L1 与 EGFR 在胶质母细胞瘤 (GBM) 中的密切关系,设计合成并筛选 EGFR/PD-L1 双重抑制剂 EP26。EP26 对 EGFR (IC50= 37.5 nM) 和 PD-1/PD-L1 相互作用 (IC50 = 1.77 μM) 表现出最高的抑制活性,并表现出优异的体外抗增殖活性和体外免疫调节作用[21]。同时,EP26 具有良好的药代动力学特性 (F = 22%),并且在 GBM 小鼠模型中抑制肿瘤生长 (TGI = 92.0%) 的效果优于Gefitinib (77.2%) 和 NP19 (82.8%)。此外,EP26 增加了肿瘤微环境中的 CD4+细胞和 CD8+ 细胞。EP26 是首个基于小分子的 PD-L1/EGFR 双重抑制剂,值得进一步研究作为癌症治疗的免疫调节剂。
图 8. EP26 显著抑制 EGFR/ PD-L1 靶点活性并具有体内抗肿瘤活性[21]。 PD-L1/CD73 抑制剂
CD73 是一种在肿瘤免疫微环境中起到关键作用的酶,通过催化 AMP 转化为腺苷,进而影响肿瘤微环境中的免疫反应。已有的临床前研究表明,靶向 CD73 的抗体或基因敲除 CD73 可以有效阻断肿瘤生长和转移,且 CD73 抗体与 PD-1/PD-L1 抗体联合使用可能产生协同效应[22]。
Wang 等设计并合成一系列双功能 PD-L1/CD73 小分子抑制剂。其中,PD-L1/CD-73-IN-1(CC-5) 表现出最强的 PD-L1 抑制效果,IC50 为 6 nM,并表现出强效的抗 CD73 活性,IC50 为 0.773 μM[23]。
SPR 实验进一步证实 CC-5 具有较高的 PD-L1/CD73 抑制活性,对人 PD-L1 和CD73 的 KD 分别为 182 nM 和 101 nM。CC-5 在 CT26 和 B16-F10 肿瘤模型中显著抑制肿瘤生长,TGI 分别为 64.3% 和 39.6%。肿瘤浸润淋巴细胞的免疫组织化学和流式细胞术分析表明,CC-5 通过激活肿瘤免疫微环境发挥抗癌作用。总之,CC-5 是第一个双重 PD-L1/CD73 抑制剂,值得作为双功能免疫治疗剂进行进一步研究。
04
小结
随着 2024 年上半年 First-in-Class 药物的涌现,我们见证了医药创新的无限可能。这些划时代的产品不仅重塑了治疗格局,更为患者点燃了希望之光。期待未来,这些创新成果能继续拓展医疗边界,造福更多生命。
参考文献:
[1] New Drug Therapy Approvals 2023.
[2] Fang W, et al. Discovery of the First-in-Class RORγ Covalent Inhibitors for Treatment of Castration-Resistant Prostate Cancer. J Med Chem. 2024 Jan 25;67(2):1481-1499.
[3] Maw JJ, et al. Discovery and Characterization of Selective, First-in-Class Inhibitors of Citron Kinase. J Med Chem. 2024 Feb 22;67(4):2631-2666.
[4] Rooney L, Jones C. Recent Advances in ALK2 Inhibitors. ACS Omega. 2021 Aug 6;6(32):20729-20734.
[5] Katagiri T, et al. Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders. Biomedicines. 2021 Jun 26;9(7):736.
[6] González-Álvarez H, et al. Discovery of Conformationally Constrained ALK2 Inhibitors. J Med Chem. 2024 Mar 28;67(6):4707-4725.
[7] Lisa Marshall, et al. Abstract 3153: Targeting the stress response kinase GCN2 to restore immunity in the tumor microenvironment. Cancer Res 1 July 2021; 81 (13_Supplement): 3153.
[8] Thomson CG, et al. Discovery of HC-7366: An Orally Bioavailable and Efficacious GCN2 Kinase Activator. J Med Chem. 2024 Apr 11;67(7):5259-5271.
[9] Eissler CL, et al. The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol Cell. 2014 Apr 10;54(1):80-93.
[10] Dong J, et al. Development of Novel Phosphonodifluoromethyl-Containing Phosphotyrosine Mimetics and a First-In-Class, Potent, Selective, and Bioavailable Inhibitor of Human CDC14 Phosphatases. J Med Chem. 2024 Jun 13;67(11):8817-8835.
[11] Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016 Feb;22(2):128-34. doi: 10.1038/nm.4036.
[12] Xie X, et al. Small-molecule dual inhibitors targeting heat shock protein 90 for cancer targeted therapy. Bioorg Chem. 2023 Oct;139:106721. doi: 10.1016/j.bioorg.2023.106721. Epub 2023 Jul 8.
[13] Sharma S, et al. First-in-Class Dual EZH2-HSP90 Inhibitor Eliciting Striking Antiglioblastoma Activity In Vitro and In Vivo. J Med Chem. 2024 Feb 22;67(4):2963-2985.
[14] Li X, et al. Design, Synthesis, and Biological Evaluation of Hydrophobic-Tagged Glutathione Peroxidase 4 (GPX4) Degraders. Bioorg Chem. 2024 Mar;144:107115.
[15] Zhu J, et al. Design, Synthesis, and Biological Evaluation for First GPX4 and CDK Dual Inhibitors. J Med Chem. 2024 Feb 22;67(4):2758-2776.
[16] Dang L, et al. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016 Apr;27(4):599-608.
[17] Wen F, et al. Drug discovery targeting nicotinamide phosphoribosyltransferase (NAMPT): Updated progress and perspectives. Bioorg Med Chem. 2024 Feb 1;99:117595.
[18] Wen F, et al. Discovery of Novel Dual Inhibitors Targeting Mutant IDH1 and NAMPT for the Treatment of Glioma with IDH1Mutation. J Med Chem. 2024 Jun 13;67(11):8667-8692.
[19] Ma L, et al. Discovery and Proof of Concept of Potent Dual Polθ/PARP Inhibitors for Efficient Treatment of Homologous Recombination-Deficient Tumors. J Med Chem. 2024 Mar 14;67(5):3606-3625.
[20] Yi M, et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022 Jan 21;21(1):28. doi: 10.1186/s12943-021-01489-2.
[21] Yang Z, et al. Discovery of Novel Small-Molecule-Based Potential PD-L1/EGFR Dual Inhibitors with High Druggability for Glioblastoma Immunotherapy. J Med Chem. 2024 May 23;67(10):7995-8019.
[22] Yu M, et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Commun. 2020 Jan 24;11(1):515. doi: 10.1038/s41467-019-14060-x. PMID: 31980601; PMCID: PMC6981126.
[23] Wang S, et al. Discovery of Small and Bifunctional Molecules Targeting PD-L1/CD73 for Cancer Dual Immunotherapy. J Med Chem. 2024 Jun 13;67(11):9447-9464.
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2024-9-13 14:15
发表于 2024-9-13 14:15
 提升卡
提升卡


