金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
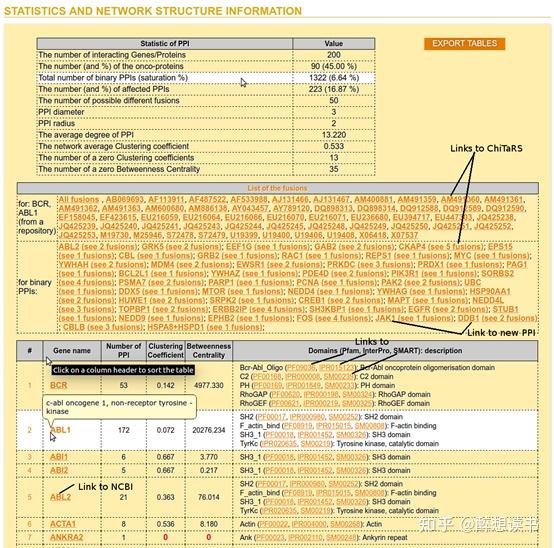
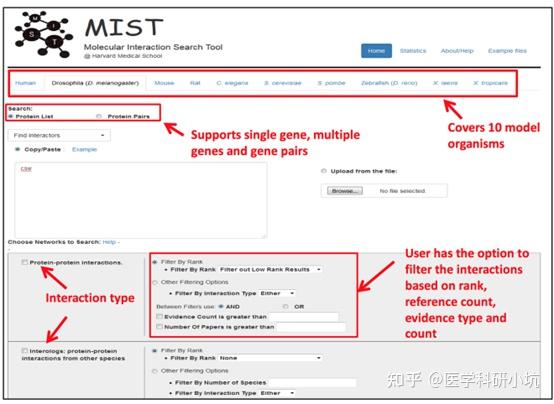
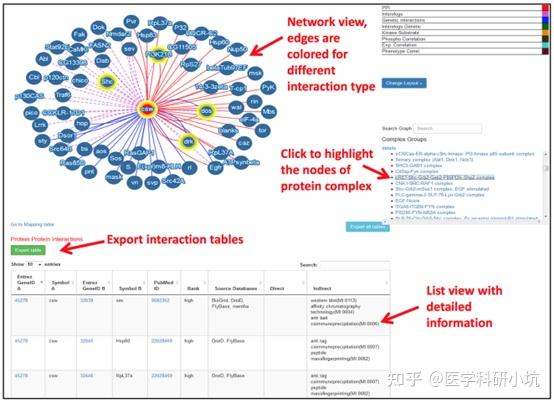
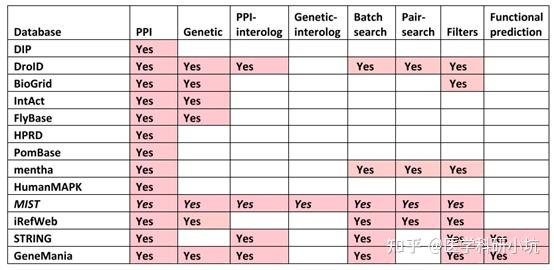
Protein-Protein Interaction,以下简称PPI
此回答针对是预测PPI复合物结构的。只要序列不是太长(单体最长1-2k),且都是20种天然氨基酸,可用AlphaFold-Multimer:
paper[1]:
Protein complex prediction with AlphaFold-Multimersource code本地版:
https://github.com/deepmind/alphafoldColab在线版:
Google Colaboratory如果没有装AlphaFold-Multimer,加了chain_break[2]的AlphaFold2[3]也有一定PPI预测能力,也可以用,Sergey提供了Colab[4]在线版:
Google Colaboratory感谢钟博子韬的评价:
精度:
AlphaFold-Multimer >> AlphaFold-Linker~AlphaFold-Gap(ColabFold)(这两个都是AlphaFold2魔改版) >> ClusPro(此前的SOTA方法)
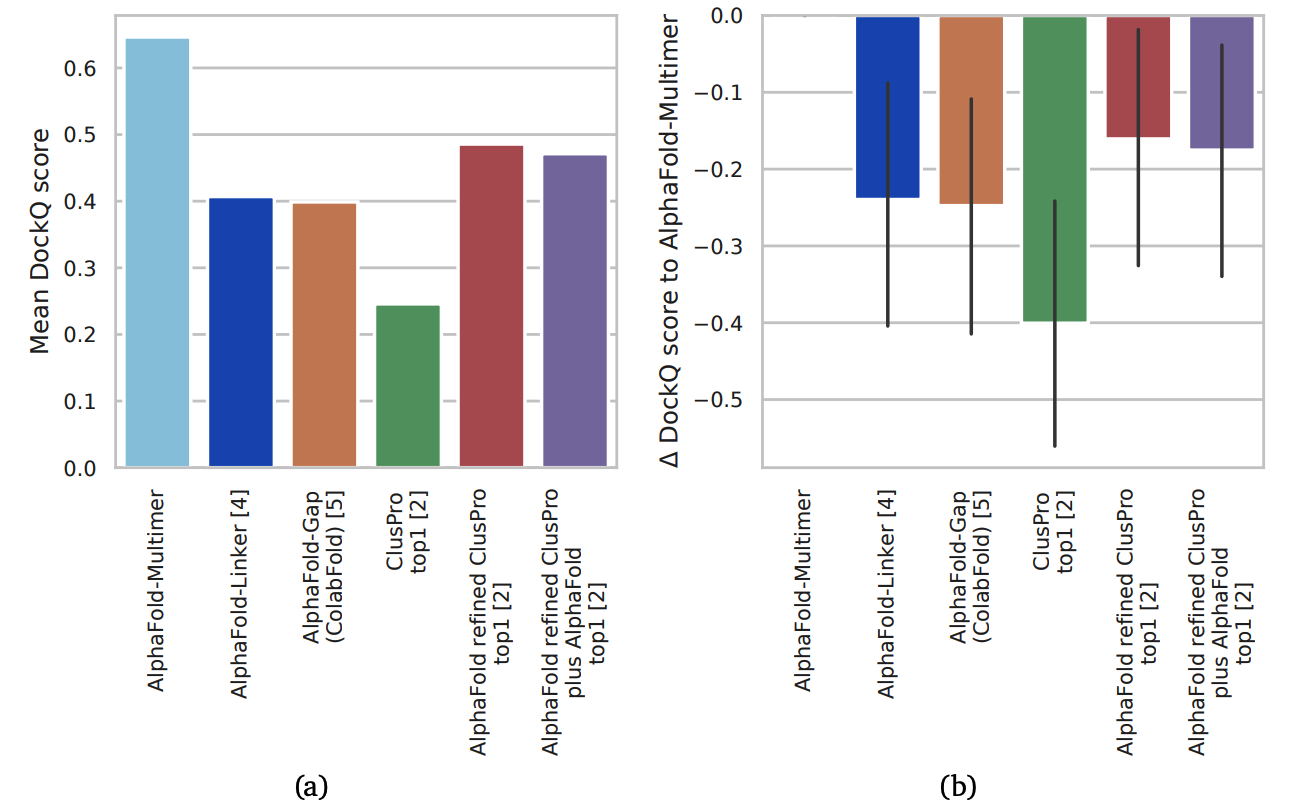
AlphaFold-Multimer文章里的原图[1]。DockQ评估PPI的预测质量,score位于[0,1],>0.8意为质量较高,<0.2基本为预测错误
适用范围:
- 同源PPI:AlphaFold2(Gap和Linker魔改版) > AlphaFold-Multimer
- 异源PPI:AlphaFold-Multimer >> AlphaFold2(Gap和Linker魔改版)
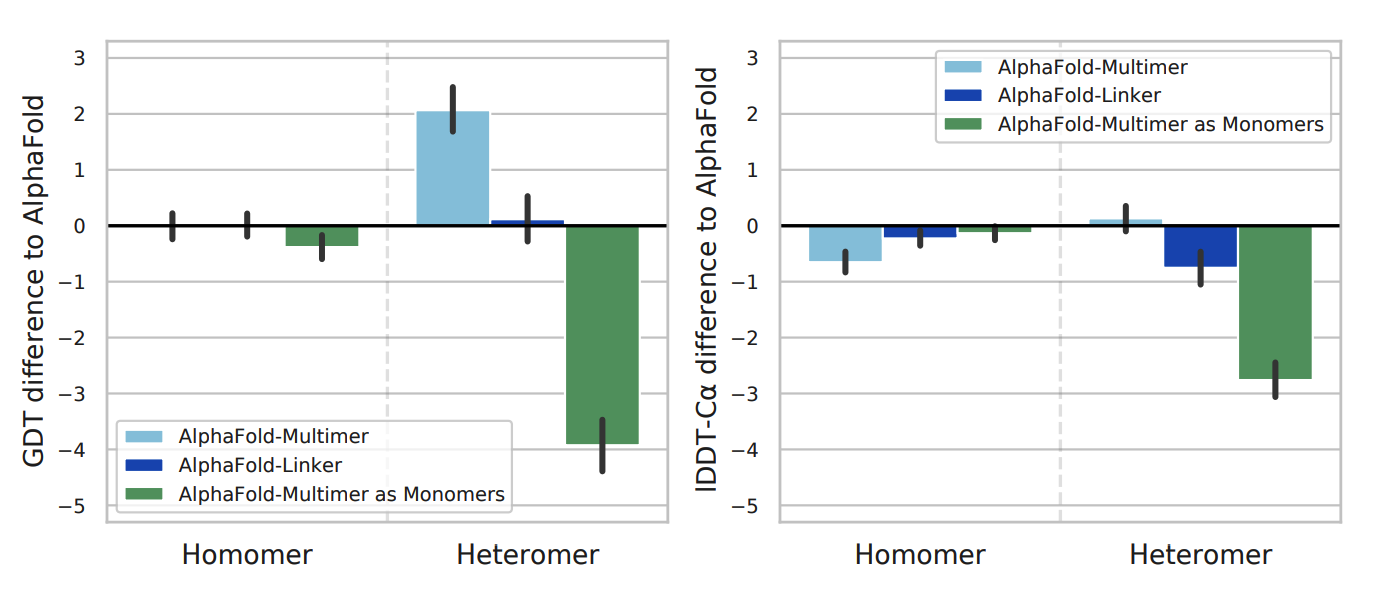
AlphaFold-Multimer文章里的原图[1]
由于AF-Multimer训练时更多的采用heteromer(异源多聚体)的complex data,在heteromer的PPI预测上比AlphaFold2(魔改版)更精确;但超参数明显没有tune好homomer(同源多聚体)上居然还开了倒车,相比AlphaFold2(魔改版)反向升级了,AlphaFold2(魔改版)训练集里存在homomer,本身就有这部分PPI预测能力。
使用建议:
- 异源PPI用AlphaFold-Multimer(v2.1.1)
- 同源PPI用AlphaFold2(v2.0.1)
- 省钱白嫖,请用ColabFold里的AlphaFold_advanced
- 批量预测,请用钟博子韬的ParaFold[5]:
About · ParaFoldReference:
Evans, Richard, et al. "Protein complex prediction with AlphaFold-Multimer."bioRxiv(2021).
Minkyung Baek (@minkbaek). Twitter post: Adding a big enough number for residue_index feature is enough to model hetero-complex using AlphaFold (green&cyan: crystal structure / magenta: predicted model w/ residue_index modification). https://twitter.com/minkbaek/status/1417538291709071362. 2021-07-20.
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873):583–589, 2021.
Sergey Ovchinnikov, Milot Mirdita, and Martin Steinegger. ColabFold-making protein folding accessible to all via google colab, 2021.
Zhong, Bozitao, et al. "ParaFold: Paralleling AlphaFold for Large-Scale Predictions."arXiv preprint arXiv:2111.06340(2021). |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-9 07:16
发表于 2024-9-9 07:16



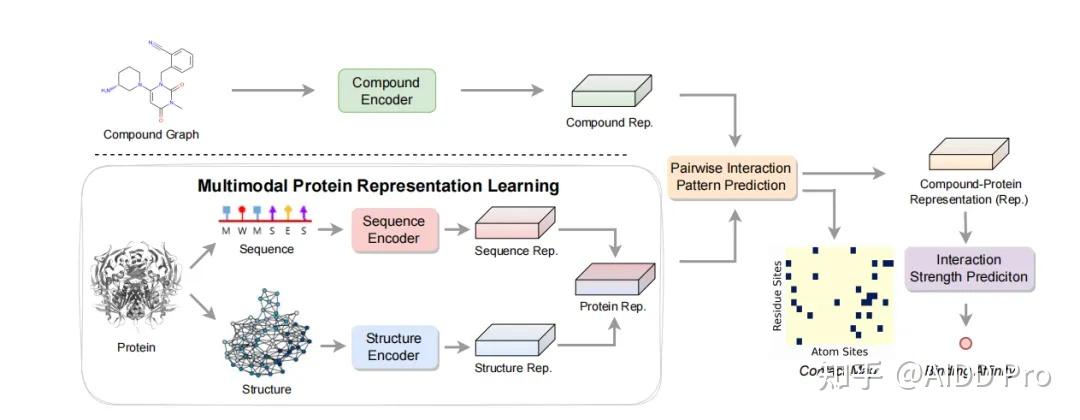
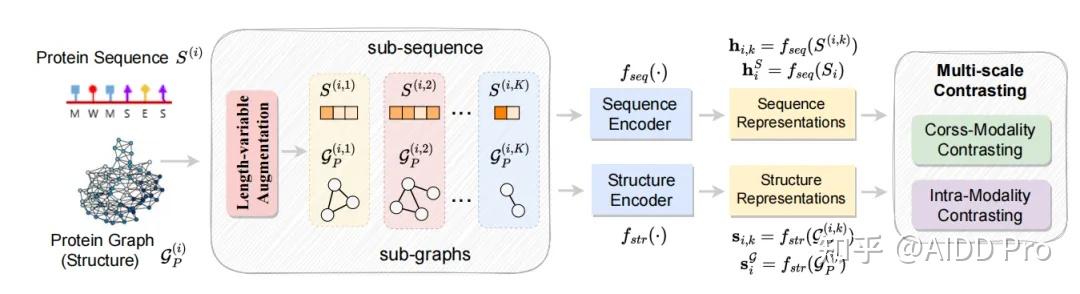

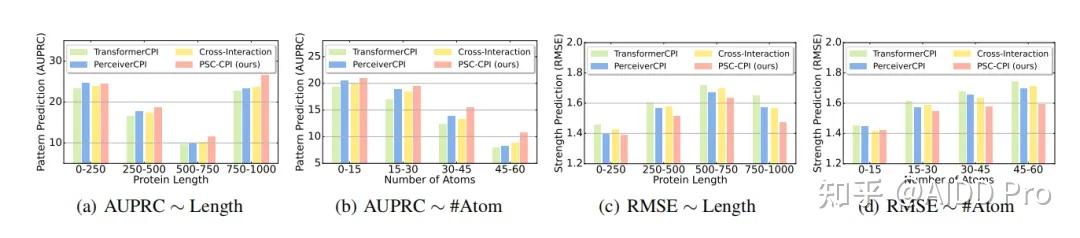
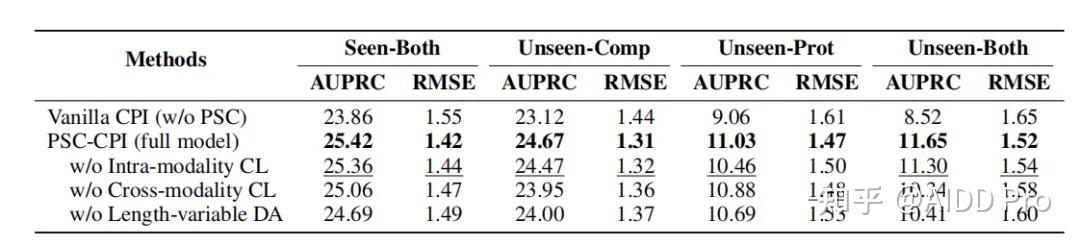
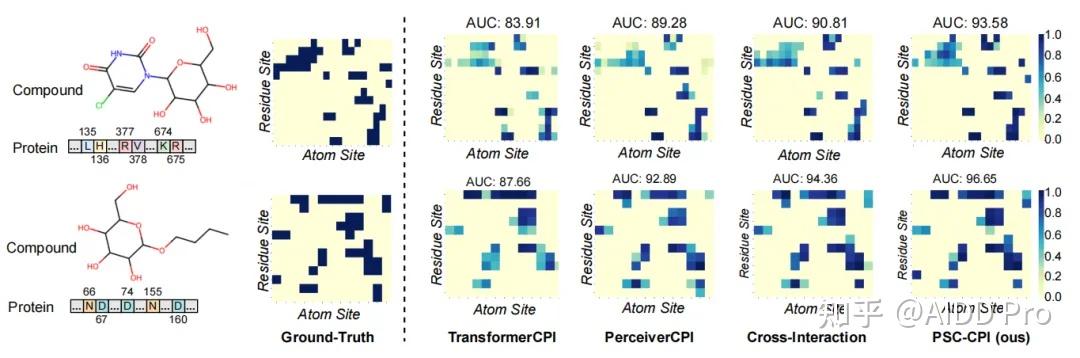




 发表于 2024-9-9 07:18
发表于 2024-9-9 07:18