金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
PCR——实验室必备实验,从基因鉴定到信号通路验证,几乎每周都要 P 个三五次,但是,有多少人能将 RT-PCR、qPCR、Real-time PCR、Real-time RT-PCR 区分开?有多少同学还在犯晕!
本文将从以下几个方面为大家详细的讲解PCR的相关基础知识内容!
- PCR原理
- 影响实验结果的关键因素
- PCR一般步骤
- 不同类型的19种PCR技术:
- PCR技术应用
最全的生物实验protocol与生物实验操作视频学习资料(细胞功能分析、蛋白组学、病例组化、分子生物学)
112个科研软件免费版(数据处理+绘图+翻译+生物实验相关等)
下载链接:https://pan.quark.cn/s/34a75dd2 一、PCR原理和历史
PCR,通常指的是普通PCR,以双链DNA为模板,以dNTP为底物,定性扩增双链DNA。
PCR(Polymerase Chain Reaction),中文全称为聚合酶链式反应,是分子生物学研究中最为广泛应用的技术。在70年代的时候首次报道使用聚合酶及合成引物进行单链DNA的扩增,但直到1983年才正式作为一种研究工具用于DNA的扩增。
1983年,美国生物化学家Kary Mullis在深夜开车回家时,突然有了灵感。他在一张收据的背面写下了最终使他在1993年获得诺贝尔化学奖的想法。这个概念很简单:在实验室的试管中复制在细胞中发生的DNA复制过程。其结果是一样的:在现有的基础上产生新的互补DNA(cDNA)链。
二、标准PCR实验原理
PCR用于从被称为模板DNA的起始材料复杂混合物中扩增出一个特定的DNA片段。样品制备和纯化方案取决于起始材料,包括样品基质和目标DNA的可及性。通常情况下,需要将DNA提纯到它的最小扩增浓度。然而,PCR确实需要了解待扩增DNA片段(称为目标DNA)侧面的DNA序列信息。
从实用的角度来看,PCR实验相对简单,可以在几个小时内完成。
影响PCR实验结果有5个关键因素:
①待扩增DNA:也叫PCR模板或模板DNA。这种DNA可以是任何来源的,如基因组DNA(gDNA)、cDNA和质粒DNA。
②DNA聚合酶:所有PCR反应都需要一种能在高温下工作的DNA聚合酶。Taq聚合酶是常用的一种,它能在70℃下以60个碱基/秒的速度结合核苷酸,并能扩增长达5kb的模板,因此它适用于标准的PCR,没有特殊要求。新一代的聚合酶正在被设计来改善反应性能。例如,有些聚合酶被设计成只在高温下激活,以减少反应开始时的非特异性扩增。其他的聚合酶则具有“校对”功能,例如,在克隆过程中,扩增序列与模板序列完全一致时,这种功能是非常重要的。
③引物:DNA聚合酶需要一个简短的核苷酸序列来指示它们需要启动扩增的位置。在PCR中,这些序列被称为引物,是单链DNA的短片段(大约15 ~ 30个碱基)。在设计PCR实验时,研究人员要确定要扩增的DNA区域,并设计一对引物,一个在正向链上,一个在反向链上,专门针对目标区域的侧翼。
引物设计是PCR实验的一个关键部分,应谨慎进行。引物序列的选择必须以感兴趣的独特DNA为目标,避免与类似序列结合的可能性。它们应该有相似的熔解温度,因为退火步骤对两条链来说是同时发生的。
如何进行引物设计?引物的熔化温度可能受到鸟嘌呤(G)或胞嘧啶(C)与腺嘌呤(A)或胸腺嘧啶(T)相比所占百分比的影响,较高的GC含量会提高熔化温度。调整引物长度可以帮助在匹配引物对时补偿这一点。避免形成二级结构或引物二聚体的序列也很重要,因为这将降低PCR效率,有许多免费的在线工具可用于帮助设计引物。
④脱氧核苷酸三磷酸酯(dNTPs):这些作为合成DNA新链的构件,包括四种基本的DNA核苷酸(dATP、dCTP、dGTP和dTTP)。dNTPs通常以等摩尔量添加到PCR反应中,以实现最佳的碱基结合。
⑤PCR缓冲液:PCR缓冲液确保在整个PCR反应中保持最佳条件。PCR缓冲液的主要成分包括氯化镁(MgCl2)、tris-HCl和氯化钾(KCl)。氯化镁作为DNA聚合酶的辅助因子,而tris-HCl和氯化钾在反应期间保持稳定的pH值。
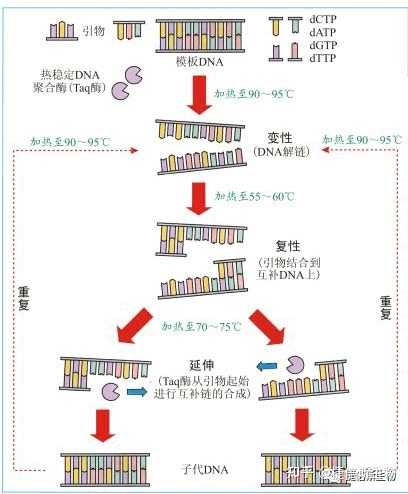
PCR的三个标准步骤
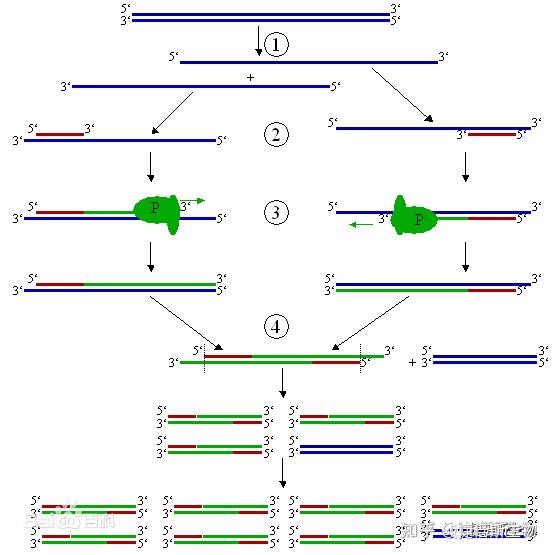
PCR可在短时间内将单个DNA扩增得到成千上万个拷贝。其过程主要由3步组成:
- 变性
PCR的第一步,称为变性,将模板DNA加热到95℃,持续几秒钟,随着两条DNA链之间的氢键迅速断裂而分离。
- 退火
然后将反应混合物冷却30秒至1分钟。退火温度通常为50 ~ 65 ℃,然而,确切的最佳温度取决于引物的长度和序列,每套新的引物都必须仔细优化。
两条DNA链可以在这个温度下重新结合,但大多数不会,因为混合物中含有大量过量的引物,它们在特定的互补位置与模板DNA结合,或退火。一旦退火步骤完成,模板DNA和引物之间将形成氢键。这个时候,聚合酶已经准备好扩展DNA序列。
- 延伸
然后将温度提高到混合物中存在的DNA聚合酶理想工作温度,通常在72℃左右,如果是Taq,则为74℃。
DNA聚合酶附着在每个引物的一端,合成新的DNA链,与模板DNA互补。现在我们有四条DNA链,而不是一开始就有的两条。
温度回升到94℃,双链DNA分子,其中包括“原始”分子和新合成的分子,再次变性为单链。这就开始了变性-退火-延伸的第二个循环。在这第二个循环结束时,有8个单链DNA分子。通过重复30次循环,开始时存在的双链DNA分子被转化为超过1.3亿个新的双链分子,每个分子都是由两个引物的退火点划定起始分子区域的副本。
为了确定扩增是否成功,PCR产物可以通过凝胶电泳进行可视化,显示扩增子的存在/不存在、大小和大致丰度。根据应用和研究问题,这可能是一个实验的终点,例如,如果确定一个基因是否存在。否则,PCR产物可能只是更复杂的下游调查(如测序和克隆)的起点。
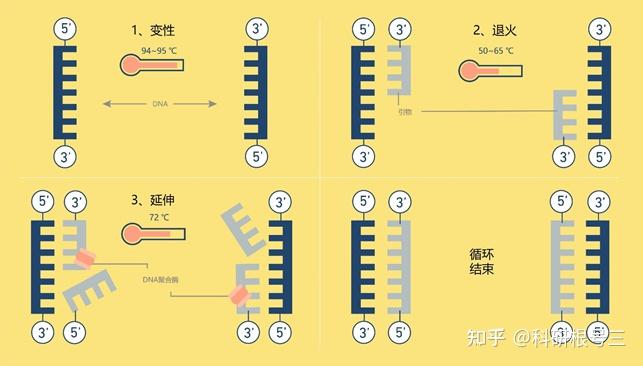
图1 | 一个PCR循环的步骤
这些步骤中的每一个都被称为循环,重复30 ~ 40次,每个循环的DNA数量翻倍,获得扩增(图2)。
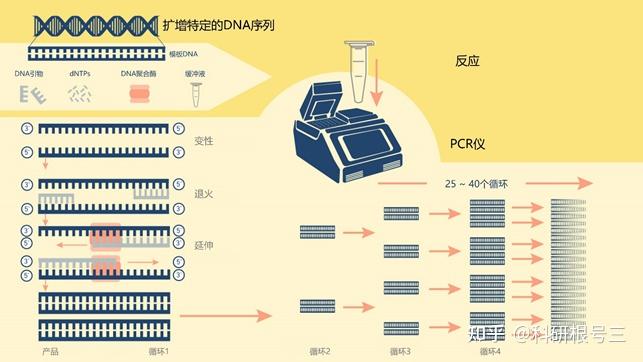
图2 | 通过PCR进行DNA分子扩增的不同阶段和循环
三、不同类型的PCR
由于其多功能性,PCR技术近年来不断发展,导致了多种不同类型的PCR技术的发展。
以下是我们可能会接触或认识到的19种PCR类型。
1、实时荧光定量PCR(Quantitative Real-time PCR, qPCR)
Real-time-PCR 和 qPCR 是一码事。实时荧光定量PCR,也叫Real-Time PCR,即二代PCR,是指在PCR扩增反应体系中加入荧光染料或者荧光基团,在整个PCR过程中通过收集荧光信号实时监测每一个循环中扩增产物量的变化,最后通过标准曲线和CT值对待测样品进行定量分析。qPCR常用的有两种方法:SYBR Green法和TaqMan探针法。
它可以用来确定目标DNA的起始浓度,在许多情况下可以忽略凝胶电泳的需要。这要归功于加入了非特异性的荧光插层染料,如SYBR® Green,它与双链DNA结合后会发出荧光,或DNA寡核苷酸序列特异性的荧光探针,如水解(TaqMan)探针和分子信标。
探针与扩增子内的DNA目标序列特异性结合,并利用福斯特共振能量转移(FRET)的原理,通过一端的荧光分子和另一端的淬灭剂的耦合产生荧光。对于荧光染料和探针来说,随着目标DNA拷贝数的增加,荧光水平也按比例增加,从而可以参照含有已知拷贝数的标准对扩增进行实时定量(图3)。

图3 | qPCR扩增图和标准曲线示例,用于对未知样本的拷贝数进行量化。
qPCR使用配备有荧光检测系统的专用PCR仪,在扩增过程中监测荧光信号。
2、反转录-PCR (RT-PCR)
反转录PCR也叫逆转录PCR。反转录(RT)-PCR和RT-qPCR是两种常用的PCR变体,能够在临床和研究环境中进行基因转录分析和病毒RNA的定量。
RT是用单链模板RNA制作cDNA的过程[3],因此也被称为第一链cDNA合成。RT-PCR的第一步是在RNA模板和DNA寡核苷酸引物之间合成一个DNA/RNA杂交体。催化这一反应的逆转录酶具有RNase活性,然后降解杂交体的RNA部分。随后,通过逆转录酶的DNA聚合酶活性合成一个单链DNA分子。高纯度和高质量的起始RNA对于成功的RT-PCR是至关重要的。
RT-PCR可以按照两种方法进行:一步RT-PCR和两步RT-PCR。在第一种情况下,RT反应和PCR反应发生在同一试管中,而在两步RT-PCR中,这两个反应是分开的,并依次进行。
注意了,虽然 Real-time PCR(实时荧光定量 PCR)和 reverse transcription PCR(反转录 PCR)看起来都可以缩写为 RT-PCR,但是,国际上的约定俗成的是:RT-PCR 特指反转录 PCR,而 Real-time PCR 一般缩写为 qPCR(Quantitative Real-Time PCR)
3、反转录-实时荧光定量PCR(RT-qPCR)
Real-time RT-PCR是qPCR和RT-PCR的组合,其中的“RT”是Reverse transcription(逆转录)的意思,所以RT-qPCR就是结合了荧光定量技术的逆转录PCR,即以mRNA或总RNA为模板,先反转录得到cDNA,再以cDNA为模板,通过荧光定量PCR进行定量检测分析。因为RT-PCR只可以定性,但不能进行定量分析。与RT-PCR一样,RT-qPCR定量分析RNA也有两种方法:一步法和两步法。两种方法都需要先将RNA反转录为cDNA,然后再将其作为qPCR扩增的模板,只是一步法中的RT和qPCR在同一试管中进行,两步法中的RT和qPCR是按顺序分开进行。
4、数字PCR(dPCR)和数字滴定PCR(ddPCR)
数字PCR即三代PCR,是对原始PCR的另一种改进,是一种能够实现核酸绝对定量的精准检测技术,基于泊松分布原理将核酸样品分配到大量独立、平行的微反应单元(纳升级)中,使每个反应室中平均只有一个拷贝或者没有目标DNA分子,然后加入荧光信号进行扩增,从而实现靶标核酸分子的绝对计数,提高检测灵敏度和准确度。
与qPCR一样,dPCR技术使用DNA聚合酶,利用引物组和探针从复杂的样品中扩增目标DNA。不过,主要的区别在于PCR反应的分区和最后的数据采集。
dPCR和ddPCR是基于限制性稀释的概念。PCR反应被分割成大量纳升体积的子反应(分区)。PCR扩增是在每个液滴内进行的。在PCR之后,用泊松统计学对每个液滴进行分析,以确定PCR阳性液滴在原始样品中的百分比。
一些分区可能包含一个或多个目标拷贝,而其他分区可能不包含目标序列。因此,分区分类为阳性(检测到目标)或阴性(未检测到目标),为数字输出格式提供基础。
ddPCR是最近的一项技术,在2011年开始使用。ddPCR利用水油乳剂形成分隔模板DNA分子的分区。这些液滴基本上充当独立的试管,PCR反应就在其中进行。
5、微流控PCR
带有微通道和微室的微流控处理系统最近的发展为一系列的实际应用铺平了道路,包括在微流控芯片上通过PCR扩增DNA。
在芯片上进行的PCR得益于微流控技术在速度、灵敏度和试剂低消耗方面的优势。这些特点使微流控PCR对POCT特别有吸引力,例如,用于诊断应用。从实用的角度来看,样品流经一个微流控通道,反复通过反映PCR不同步骤的三个温度区。10 μL的样品进行20个PCR循环只需要90秒[6]。随后的分析可以很容易地在片外进行。
6、降落PCR(touchdown PCR)
降落PCR,主要用于PCR的条件的优化。在许多情况下引物的设计使得PCR难以进行,例如特异性不够易错配等。退火温度过高会使PCR效率过低,但退火温度过低则会使非特异扩增过多。
因此,前面几个循环的起始退火温度设定为比引物的最高熔解温度(Tm)再高几度。前几循环温度逐渐下降至设定的最终Tm。通过较高温度获得特异性匹配较高的模板后,再以较低温度高效率扩增。
7、巢式 PCR(nested PCR)
巢式 PCR,先用低特异性引物扩增几个循环以增加模板数量,再用高特异性引物扩增。巢式PCR是常规PCR的一种演变,其增强了反应特异性和目标扩增子的产量。
巢式 PCR,通过使用两套引物来进行两次连续的反应,用来增加 DNA 扩增的特异性。在第一次反应中产生的产物可能包含非特异性扩增产物,然后使用两个新的、结合位点位于原引物内部的引物进行第二次反应。第二套引物的使用可提高反应的特异性,增大单一产物产生的可能性。巢式 PCR 在提高特异性的同时,也增加了检测的灵敏度。

8、高GC含量PCR
具有高GC含量(>65%)的DNA模板由于G和C碱基间的强氢键影响,比较难以扩增。富含GC的序列同时也涉及二级结构。因此,富含GC的序列可导致DNA聚合酶沿模板扩增时“卡顿”并干扰DNA合成。
为了扩增高GC含量的片段,双链模板必须解离,以便引物与模板结合,并使DNA聚合酶能够读取到序列。为了克服强GC相互作用,最常用的方法是使用DMSO等PCR添加剂或辅助溶剂来帮助DNA变性。然而,这些试剂通常会降低引物的 Tm,所以退火温度也需进行相应的调整。
高合成能力的DNA聚合酶由于与模板的结合能力更强,有利于完成高GC含量PCR。超高热稳定性DNA聚合酶也有利于高GC含量PCR,因为较高的变性温度(如,使用98°C代替95°C)可能会促进双链解离和PCR扩增。
9、等位基因特异性PCR(AS-PCR)
等位基因特异性PCR( allele specific PCR,AS-PCR ),是指利用引物与模板之间的碱基错配可以有效地抑制PCR反应,进而达到模板区分(等位基因区分)的目的。
由于PCR过程中引物延伸是3'端开始的,所以3'末端的碱基对引物的延伸来说处于至关重要的位置。如果这个碱基与模板互补,则引物能不间断延伸,PCR可以正常进行,得到特定长度扩增带,反之,则不能延伸。所以只要将与正常等位基因所不同的那个突变碱基安排在引物3'最末端,当用某一含突变序列的引物进行PCR时,如果得到特异条带,表明被测基因含有该种突变。没有特异扩增带出现,则表示没有这种突变。
注意:这里由于仅仅利用了引物3'末尾碱基的错配,因此需要摸索一个合适的Tm,才能达到检测目的。
10、多重 PCR(Multiplex-PCR)
多重 PCR,指在一个 PCR 管中使用多对引物同时扩增超过一个的靶基因。同时分析单拷贝基因组的多个基因可避免分别扩增这些靶基造成对试剂和模板的浪费。使用多重 PCR 检测引起遗传疾病的基因突变时,可以同时扩增 6 个以上的靶点,也可同时检测食品中多种病原菌的存在。在多重 PCR 基础上发展的 Multiplex Ligation-dependent Probe Amplification(MLPA,多重连接探针扩增技术) 使用单对引物即可完成对多个靶基因的扩增,避免了多重 PCR 耗时的优化步骤。
11、不对称 PCR(Asymmetric PCR)
不对称 PCR,可选择性的扩增出靶 DNA的一条链,从而用于测序反应或制备单链杂交探针。反应中限制一个引物的加入量,随着这一引物的用尽,后续的扩增中另一引物延伸的产物量大大过量。这一技术的关键是使用的限制引物的量要合适,引物量过多,则产物主要是双链 DNA;引物量过少,在前几轮循环就被耗尽,结果导致单链的产量减少。在不对称 PCR 的基础上发展出 Linear-After-The-Exponential-PCR (LATE-PCR,线性指数 PCR),使数量限制的引物比过量的引物的解链温度更高,从而维持较高的反应效率。
12、反向 PCR(Inverse PCR)
反向 PCR,允许扩增已知序列侧翼的 DNA 区。首先将靶 DNA 用限制性内切酶进行多轮消化,然后连接被消化的片段构建环状的分子,引物设计成可从序列已知的区域向外侧延伸,结果扩增出环状分子上剩余的序列。
13、锚定PCR(Anchored PCR)
用酶法在一通用引物反转录cDNA 3’-末端加上一段已知序列, 然后以此序列为引物结合位点对该cDNA进行扩增, 称为APCR。类似RACE。它可用于扩增未知或全知序列, 如未知cDNA的制备及低丰度cDNA文库的构建。
14、原位PCR(in situ PCR)
原位PCR是一种通过在单细胞或组织切片上对特异的核酸序列进行原位扩增、检测定位的新技术。结合了具有细胞定位能力的原位杂交和高度特异敏感的PCR技术的优点,是细胞学科研与临床诊断领域里的一项有较大潜力的新技术。用于外源性基因片段检测、、观察病原体在体内分布规律、内源性基因片段检测、导入基因检测、遗传病基因检测等等。
15、重组PCR(recombinant PCR)
又叫重叠延伸PCR、overlap PCR。是指把两个不相邻的DNA片段重组在一起的PCR法。其基本原理是两个基因片段设计在引物中,先分段对模板扩增,除去多余的引物后,将产物混合,再用一对引物对其进行PCR扩增。所得到的产物是一重组合的DNA。
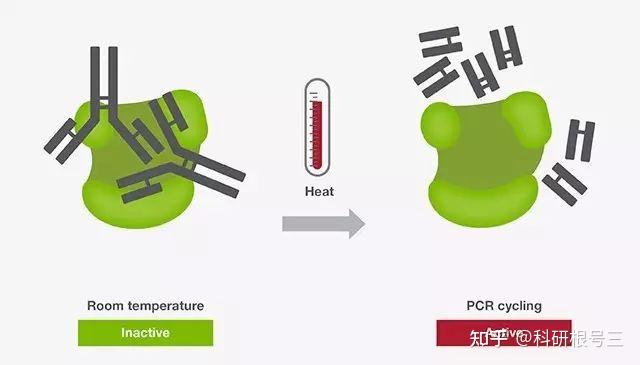
16、热启动PCR(hot start PCR)
热启动PCR常用于增强PCR扩增的特异性。热启动PCR主要借助一种酶的修饰物(如抗体、亲和配体、适体或化学修饰物)来抑制常温下DNA聚合酶的活性。这种修饰使得在PCR反应体系配制阶段引物与模板、引物与引物之间的结合能力降低,从而避免了非特异性扩增。由于DNA聚合酶在常温下的活性被抑制,热启动技术为在常温下配制多个PCR反应体系提供了极大的便利,而无需牺牲PCR反应的特异性。
当反应体系配制好后,在反应初始加热步骤,酶修饰物在高温下(通常高于90 ℃)被释放,使得DNA聚合酶被激活(图1)。激活时间和温度根据DNA聚合酶及热启动修饰物的不同而有所差别。对于某些聚合酶,激活和预变性合并成了一步。
热启动酶的出现大大提高了PCR扩增的特异性。在点突变、基因工程、基因改造等方面表现尤为突出。
17、快速PCR(Fast PCR)
在快速PCR中,通过缩减PCR步骤所需的时间来完成更快的扩增,而不影响扩增产量和效率。快速循环条件尤其适用于均有高扩增能力的DNA聚合酶,这种聚合酶在每次结合中可引入更多的核苷酸。
18、直接PCR(Direct PCR)
直接PCR意指直接从样本中扩增目的DNA,而无需核酸纯化。直接PCR中,在高温变性阶段,诸如细胞、组织等材料在独特的缓冲液中裂解,释放出DNA。因此这种方法简化了PCR实验流程,减少了动手操作的时间,同时可避免纯化步骤中DNA的损失
19、长片段PCR技术(long-PCR)
顾名思义就是进行长片段扩增的技术。此技术主要用于基因组扩增。从几kb到几十kb。此技术对于模板、酶、引物等各方面要求很高。近年来出现许多商品化的长片段扩增酶[1]。
四、PCR的应用
PCR已经成为现代分子生物学中不可缺少的工具,并完全改变了科学研究。该技术还为分子生物学领域以外的人打开了细胞和分子过程的研究,因此也被许多学科的科学家发现了用途。
虽然PCR本身是一种强大的独立技术,但它也被纳入了更广泛的技术,如克隆和测序,作为这些工作流程中一个小而重要的部分。
PCR的研究应用包括:
1、基因表达
PCR可以检查特定时间点上细胞类型、组织和生物体之间基因转录的变化。在这个过程中,RNA从感兴趣的样品中分离出来,并反转录成cDNA。然后,特定基因的原始RNA水平可以从PCR中扩增的cDNA数量中进行量化。
2、基因分型
PCR可以检测特定细胞或生物体的等位基因的序列变化。一个常见的例子是对转基因生物体进行基因分型,如基因敲除和基因敲入小鼠。在这种应用中,引物被设计用来扩增转基因部分(在转基因动物中)或突变部分(在突变动物中)。
3、克隆和诱变
PCR克隆是一种广泛使用的技术,通过PCR扩增的双链DNA片段被插入载体(如gDNA、cDNA、质粒DNA)。例如,这使得创建的细菌菌株的遗传物质被删除或插入。定点诱变也可用于通过克隆引入点突变。这通常采用一种被称为重组PCR的技术,其中重叠的引物被专门设计来纳入碱基替换(图4)。这种技术也可用于创造新的基因融合。
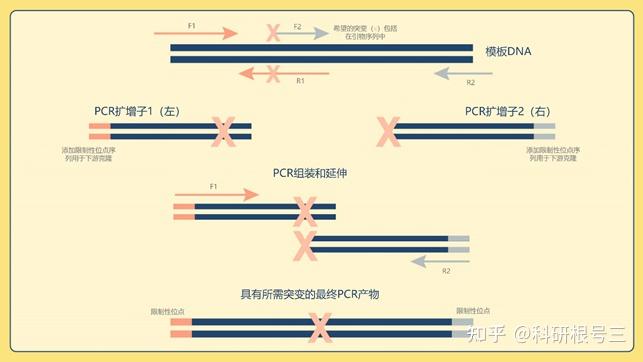
图4 | 描述重组PCR实例的图表
4、测序
PCR可用于富集测序用的模板DNA。推荐用于制备测序模板的PCR类型被称为高保真PCR,能够保持DNA序列的准确性。在Sanger测序中,PCR扩增的片段随后被纯化并在测序反应中运行。在第二代测序(NGS)中,PCR被用于文库制备阶段,通过PCR富集DNA样本以增加起始数量,并用测序适配体标记以实现多重检测。桥式PCR也是第二代NGS测序过程的一个重要部分。
5、甲基化
PCR可用于研究位点特异性甲基化。在 甲基化特异性PCR(MSP)方法中,设计了两个引物对,以区分目标位点的甲基化状态。
首先使用重亚硫酸盐处理DNA样品,将未甲基化的胞嘧啶(C)转化为尿嘧啶(U)。重亚硫酸盐处理不会影响甲基化的胞嘧啶(m5C) 。为了检测甲基化位点,一对引物经设计 带有 鸟嘌呤(G),可与目标序列中的 m5C 配对;为了检测未甲基化位点,另一对引物带有腺嘌呤(A),可与重亚硫酸盐转化分子中的U配对(随后,与后续PCR循环中的胸腺嘧啶(T)配对)。通过引物配对得到的阳性PCR扩增结果可用于确定位点的甲基化状态
6、医学、法医学和应用科学
PCR技术不仅可用于基础研究,还适用于日常的临床诊断、法医学调查和农业生物技术研究。这些应用要求可靠的性能、卓越的灵敏度和严格的标准。因此,所使用的PCR仪和PCR试剂必须符合这些要求和目的。
分子诊断应用包括基因检测、致癌突变检测以及感染性疾病检测。在法医学中,利用 PCR进行人类身份鉴定是通过对独特的短串联重复序列(STR)进行扩增而区分个体的。在农业学中,PCR在食物病原体检测、育种植物基因分型和 GMO测试中具有重要作用。
参考文献
[1] Chien A, Edgar DB, Trela JM. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J Bacteriol 1976;127(3):1550-57 doi: 10.1128/JB.127.3.1550-1557.1976
[2] Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985;230(4732):1350 doi: 10.1126/science.2999980
[3] Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel HRH. Basic principles of real-time quantitative PCR. Expert Review of Molecular Diagnostics 2005;5(2):209-19 doi: 10.1586/14737159.5.2.209
[4] Bachman J. Chapter Two - Reverse-Transcription PCR (RT-PCR). In: Lorsch J, ed. Methods in Enzymology: Academic Press, 2013:67-74. doi : 10.1016/B978-0-12-420037-1.00002-6
[5] Morley AA. Digital PCR: A brief history. Biomol Detect Quantif 2014;1(1):1-2 doi: 10.1016/j.bdq.2014.06.001
[6] Taylor SC, Laperriere G, Germain H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: from variable nonsense to publication quality data. Scientific Reports 2017;7(1):2409 doi: 10.1038/s41598-017-02217-x
[7] Ahrberg CD, Manz A, Chung BG. Polymerase chain reaction in microfluidic devices. Lab on a Chip 2016;16(20):3866-84 doi: 10.1039/C6LC00984K
[8] Garibyan L, Avashia N. Polymerase chain reaction. J Invest Dermatol 2013;133(3):1-4 doi: 10.1038/jid.2013.1
VanGuilder HD, Vrana KE, Freeman WM. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 2008;44(5):619-26 doi: 10.2144/000112776
推荐内容
绝密!2023年国自然立项清单表可下载(生命科学部+医学科学部),附往年1000+份中标标书!
如何查看国家自然科学基金的的摘要和下载结题报告?
有哪些好用的zotero插件?
有什么适合药学/生物方向科研小白的简单易学的科研绘图工具?
有没有什么很好的科研作图软件?
流式数据分析标杆神器,FlowJo 10 分享!
顶级图像分析软件,Image J、Fiji、Image pro plus,一次帮你搞定!
NoteExpress和EndNote相比哪一个更好用?(附安装包)
2023年更新!EndNote X9永久激活版本(序列号激活),最稳定,可汉化!
写的太全了,Endnote 插入文献以及调整参考文献格式,这是知乎上最好的教程!
太牛了!Zlibrary回归,这里有最稳定的访问指南!
western blot是为了什么?
如何学习和记忆细胞信号通路?
最值得推荐装!GraphPad Prism 9.3 ,功能更多!
SPSS 27 安装激活,授权日期到期时间2037年 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-9-6 21:37
发表于 2024-9-6 21:37


 发表于 2024-9-6 21:38
发表于 2024-9-6 21:38