登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
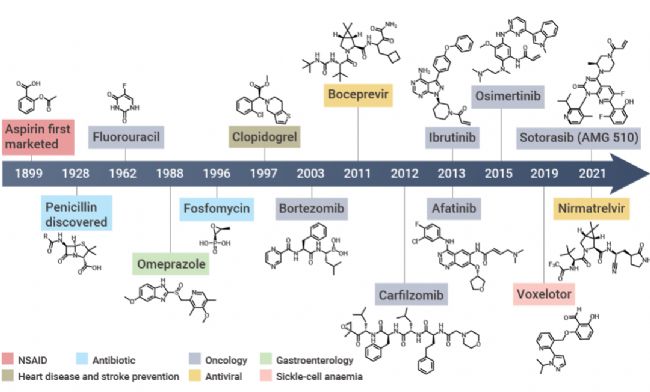
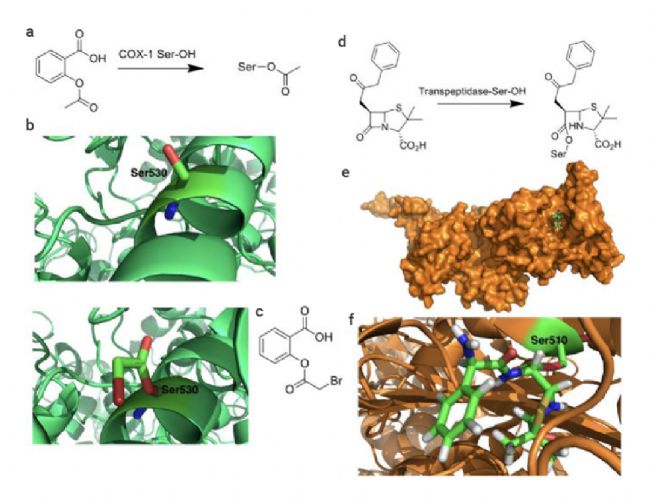
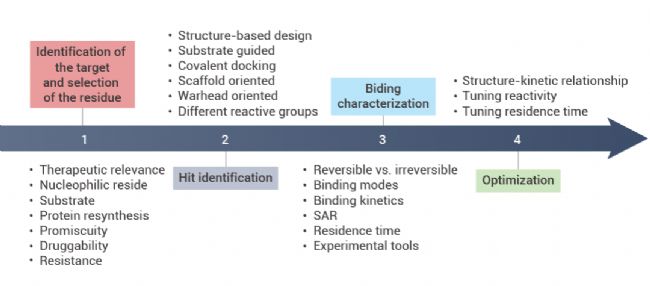
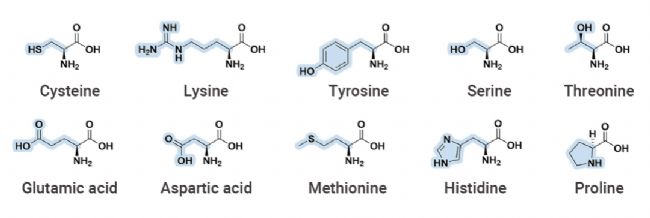
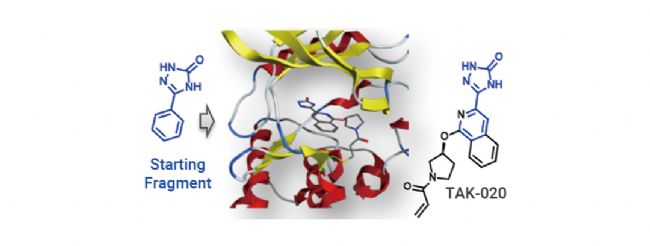
共价抑制剂最早可追溯到 17 世纪末的非甾体抗炎药 Aspirin (阿司匹林)。相较于非共价药物,共价药物的优势在于能够提高药物效力,延长靶向结合时间,选择性更强,同时可以靶向某些“不可成药” 靶点,解决耐药性问题。 越来越多的研究表明,许多已上市的药物都是通过共价作用发挥其药效,近年来一些共价蛋白激酶抑制剂的成功上市,如 Ibrutinib (BTK 共价抑制剂) 和 Dacomitinib (EGFR 共价抑制剂),更是让共价药物成为了药物开发中一个备受关注的领域[1]。 图 1. 共价药物的开发历程[1]。 在过去的共价药物的研究历史中,共价药物主要是来源自偶然发现。尽管最早的共价药物阿司匹林上市于 1899 年,但直到 1971 年阿司匹林的共价作用机制才被揭开。 阿司匹林中的活性成分乙酰水杨酸共价结合环氧合酶 (COX-1 和 COX-2),将其乙酰基转移到 COX 的丝氨酸侧链的羟基上,抑制花生四烯酸(环氧合酶的底物)转化为前列腺素,以发挥抗炎作用。青霉素类抗生素是另一类共价抑制剂,后面被发现其机制是基于青霉素 β-内酰胺类化合物与青霉素结合蛋白 1B 的活性位点丝氨酸共价结合[2]。 图 2. (a-c) 阿司匹林和 (d-e) 青霉素的共价作用机制图[2]。 由此可见,之前的共价药物都不是刻意设计的,大多数来自于天然产物,其共价机制更是在临床上被广泛使用后才被阐明。由于共价化合物在过去被认为可能带来脱靶效应,并且诱发一些免疫反应从而存在潜在的安全问题,因此当时大多数医药公司都会优先考虑非共价化合物,直到后期越来越多药物的共价机制被披露,共价化合物才开始被药物开发人员重视起来。迄今为止,共价抑制剂更是占据了上市药物约 30% 的比例。 传统的小分子药物设计是基于抑制或激活生物靶标的生物活性从而发挥作用。无论小分子药物的功能如何,其作用通常取决于活性物质 (如抑制剂、效应剂或激活剂) 与生物靶标 (如酶、蛋白质、离子通道或受体) 之间的相互作用。  Tips:共价药物与靶标发生共价结合主要分为两个过程: Tips:共价药物与靶标发生共价结合主要分为两个过程:(1) 共价药物的亲电弹头通过靠近生物靶标的活性氨基酸残基,与生物靶标可逆性结合。(2) 共价药物与生物靶标发生反应以形成共价键。 根据反应的可逆性,共价药物可以被分为可逆型共价药物和不可逆型共价药物。可逆性共价药物在设计上旨在提高药物的安全性和选择性,而不可逆性共价药物则提供了更持久的药理作用,但需要更仔细地考虑其潜在的脱靶效应和毒性风险,建议大家谨慎选择哦~ 丙烯酰胺、β-内酰胺等亲电弹头是共价药物发挥作用的结构基础,亲电弹头能与靶蛋白中特定的氨基酸残基 (如半胱氨酸、丝氨酸或苏氨酸等) 发生化学反应,形成共价键,从而抑制靶标蛋白的生物学功能。目前为止,已有几十种的亲电弹头被发现,其中在上市和待批准药物中以“丙烯酰胺类弹头”开发的共价药物数量最多[3]。 生物靶点的识别和验证是药物发现的第一步,接下来是对鉴定出的苗头化合物进行表征和结构优化。共价药物发现中使用的许多方法与传统药物化学中使用的方法相似,包括高通量筛选、虚拟筛选和合理的药物设计等。然而,在共价药物的设计中,很多地方都是独一无二的 (如靶点选择、亲电弹头和氨基酸残基的选择等)。因此,共价药物的设计一般包含四个过程:靶标识别和氨基酸残基选择;苗头化合物的识别;结合表征;结构优化[4]。 图 3. 共价药物设计的阶段图[4]。 ▐ 靶标识别及氨基酸残基的选择治疗靶标的识别是共价药物设计的第一步,往往基于遗传学、生物物理学、化学物理学或其他原理的方法进行。通常,疾病靶点应当作为药物设计的第一指导原则,在开展药物开发工作前,确认疾病-靶点的关联度非常重要。目前,基于活性的蛋白质分析 (Activity-based protein profiling, ABPP) 是用于共价化合物靶点发现最通用的化学蛋白质组学方法之一,通过反应性共价片段来确定共价作用的可能靶点。初步筛选出可能的靶点后,还需要对药物靶标进行验证,目的是通过调节靶标来验证其对疾病的治疗作用,为靶点的“二次鉴定”提供依据。 在共价药物的设计中,氨基酸残基的选择同样尤为重要。在设计共价药物时必须考虑共价弹头和氨基酸残基的反应性。此外,还需要考虑该生物靶点是否适合共价机制,以避免潜在的药物毒性,同时还需要满足药物的高选择性。在各类氨基酸残基中,半胱氨酸是最为常见的共价氨基酸残基,其代表药物有奥美拉唑、氯吡格雷、阿法替尼等[5]。 图 4. 常见的共价氨基酸残基结构[4]。 ▐ 苗头化合物的识别高通量筛选 (High-throughput screen,HTS) 是药物设计中常用技术之一,用于从化合物库中发现可能成为药物化学优化的先导化合物,可以加速大批量化合物的筛选进程。对于已知共价抑制剂及具有共价反应基团的活性小分子化合物,可以直接使用 HTS 对其进行药物筛选。 在共价药物设计中,基于片段的药物发现 (Fragment-based Drug Discovery,FBDD) 是一种较为广泛应用的方法。FBDD 适用于筛选分子量更小、结构不太复杂的分子,通过筛选出有活性的片段后,将不同片段进行组合延伸以到新的药物分子。由于片段化合物库范围更广,因此筛选出活性化合物的概率更大。并且片段具有分子量小、无效基团少的特点,后期更有利于进行结构优化[6]。例如 BTK 抑制剂 TAK-020 (一种临床前候选药物) 就是基于 FBDD 发现的[7]。 图 5. 基于 FBDD 的临床候选药物 TAK-020[7]。 此外,DNA 编码化合物库 (DNA Encoded compound Library,DEL) 技术也是一种共价药物的筛选方法,与 HTS 和 FBDD 相比,DEL 最显著的优势在于能够筛选更大的文库,可达到千万甚至数十亿级别的数量[8]。在 2020 年的疫情期间,Rui Ge 等就基于 DEL 技术,构建了具有 6 亿个化学结构的 DNA 编码文库,针对冠状病毒中的半胱氨酸蛋白酶 Mpro 进行了共价抑制剂的筛选[9]。 ▐ 结合表征药物的表征是药物发现过程中的关键步骤,传统的表征参数包括解离常数、IC50、EC50 等。而共价药物的主要表征方法有:体外生物活性测定、质谱分析、时间分辨荧光共振能量转移技术 (TR-FRET)、表面等离子体共振 (SPR) 技术等。事实上,表征参数及表征方法的选择不能一概而论,需要根据药物的本身特性去进行选择。 ▐ 结构优化在确定了苗头化合物后,我们往往需要对化合物的结构进行进一步优化,目的是改善其药物代谢动力学性质,增强共价弹头的选择性,避免脱靶效应,增强药物的作用时间等。最终经过结构优化后,就可以得到先导化合物。  在过去的十年中,共价药物在新药开发领域取得了显著进展,这主要得益于对共价作用机制的深入理解以及药物开发技术的持续创新。  [1] Boike L, et al. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21(12):881-898. [2] Ghosh AK, et al. Covalent Inhibition in Drug Discovery. ChemMedChem. 2019 May 6;14(9):889-906. [3] Ray S, et al. New Electrophiles and Strategies for Mechanism-Based and Targeted Covalent Inhibitor Design. Biochemistry. 2019;58(52):5234-5244.[4] Zheng L, et al. Development of covalent inhibitors: principle, design and application in cancer. MedComm – Oncology. 2023; 2:e56. [5] Huang F, et al. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules. 2022;27(22):7728.[6] Bon M, et al. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol Oncol. 2022;16(21):3761-3777.[7] Sabat M, et al. Discovery of the Bruton's Tyrosine Kinase Inhibitor Clinical Candidate TAK-020 (S)-5-(1-((1-Acryloylpyrrolidin-3-yl)oxy)isoquinolin-3-yl)-2,4-dihydro-3H-1,2,4-triazol-3-one, by Fragment-Based Drug Design. J Med Chem. 2021 Sep 9;64(17):12893-12902. [8] Li L, et al. Triazine-Based Covalent DNA-Encoded Libraries for Discovery of Covalent Inhibitors of Target Proteins. ACS Med Chem Lett. 2022;13(10):1574-1581. [9] Ge R, et al. Discovery of SARS-CoV-2 main protease covalent inhibitors from a DNA-encoded library selection. SLAS Discov. 2022 Mar;27(2):79-85.
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2024-5-24 15:02
发表于 2024-5-24 15:02