金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×
分享|CADD之分子动力学的简介(下篇)在上篇中,我们了解了分子动力学的基础和基本原理,接下来我们来看一下MD模拟是如何保证准确性的,以及它的主要流程。
MD的准确性如何?
通常一种理论方法的出现,会伴随各种声音的出现,MD模拟也毫无例外,有支持派,当然也有质疑其准确性的。那它对分子结构和原子间相互作用描述的到底准确吗?
在上篇当中提到的范德华相互作用,其是通过LJ势进行描述的,该势中除了有与位置坐标相关的r之外,LJ势中还含有两个参数:σ和ε。以AMBER99SB力场为例,在该力场下脂肪族中的C和羰基中的C,这两个参数的值会不同。当然在不同的力场下,同种原子的这些参数也会有差别。那么这类参数是如何得到的呢?这些参数是拟合自实验数据或量子化学的结果,是通过选定初始参数,做分子模拟,然后看模拟结果和实验值的差别,重新调整参数,继续模拟,直到达到收敛标准。这一过程属于经验描述,都是为了和实验数据相符合,这也就是为什么会出现四点水模型和五点水模型的原因。而且各种力场也会根据新的实验数据去做参数优化,使其更加准确。现如今已发展了很多分子力场,比如生物模拟常用的AMBER, CHARMM, OPLS, GROMOS,材料领域常用的CFF, MMFF, COMPASS等等。
图1是与MD模拟相关的文献发表量(从1977-2017年),并且这些文章发表在顶级期刊。这表明MD模拟在近年来已经变得越来越常见,也逐渐受到了认可。
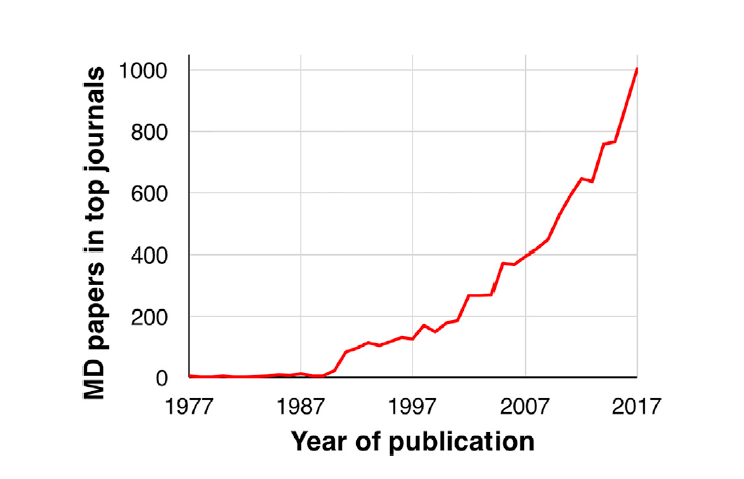
图1 与MD模拟相关的文献量
MD如何应用?
应用软件
随着计算机性能的发展,能执行MD模拟的软件也逐渐增多。现如今比较主流的MD模拟软件(见表 1),如AMBER,GROMACS,CHARMM等。当然也有一些其他可以进行MD模拟的计算软件,但由于只是该软件的一个模块,计算十分耗时,并不推荐。
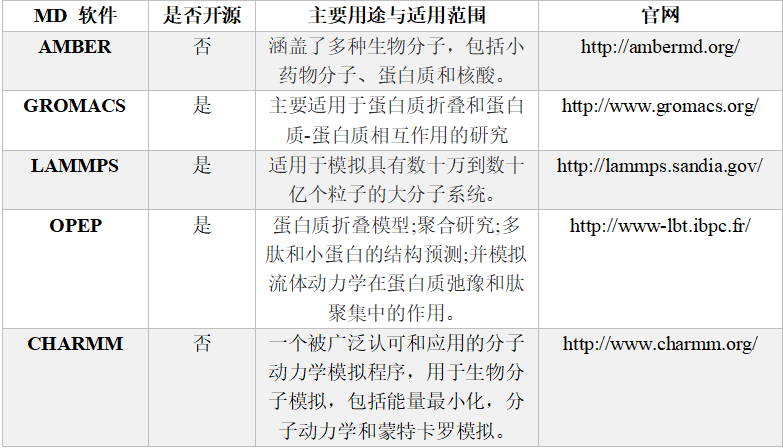
表 1 几款常用的MD模拟软件
对于初学者而言,GROMACS是相对来说较为友好的软件。这款软件是开源的,也支持GPU加速,安装简单,很容易上手。鉴于GROMACS官网上已经有很多分子动力学相关教程(包括蛋白、蛋白-小分子、蛋白-磷脂分子层、虚拟位点等),我们这里就不重复介绍已有的蛋白模拟教程,小伙伴们可以在文末找到教程网址,自行操作。
主要过程
无论是哪一种软件,它们的步骤都是相类似的,大致包含以下过程(图2):
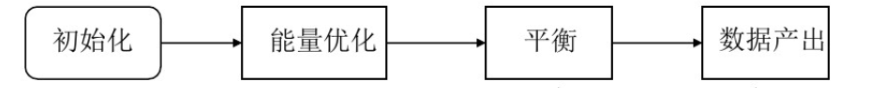
图2 主要过程
初始化
初始化是分子动力学里重要的一步,它会读取模型的参数,模拟控制参数。如增加非重原子的缺失;确定溶剂;确定周期性边界条件盒子;确定力场等。(选择恰当的力场是保证体系结果合理的前提,因此力场选择需谨慎!)
能量优化
通常有些结构模型会存在原子重叠(图3),键长和键角的严重扭曲等一些不合理的状态,若以这种状态直接进行分子动力学模拟,得到的数据也是不准确的。为了解决这一类问题,通常会对模拟体系进行能量优化,优化其模型结构。一般会优化几千到几万步。
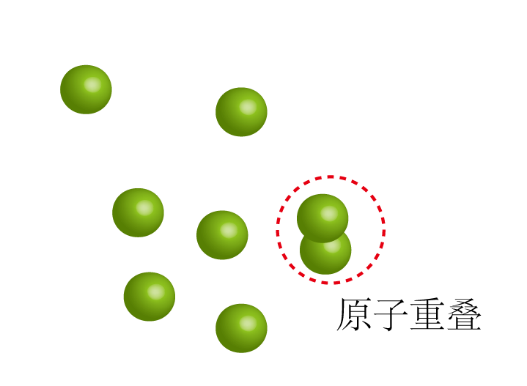
图3 原子重叠
平衡
平衡分子动力学模拟,总是在一定的系综下进行的,系综大致可分为以下几类:
(系综:代表一定条件下,一个体系的大量可能状态的集合)
微正则系统
系统原子数N,体积V,能量E保持不变。又称为 NVE系综。微正则系综里的每个体系具有同等的能量。
正则系统
系统原子数N,体积V,温度T保持不变,且总动量保持不变。又称为NVT系综。该系综里的各体系可以和其他体系进行交换能量。
等温等压系统
系统原子数N,压强P,温度T保持不变,又称为NPT系综。在等温等压系综下,各体系可以和其他体系交换能量和体积,但系综内各个体系有相同的温度和压强。
等压等焓系统
系统原子数N,压强P,焓值H=E+PV保持不变。在模拟中较少见。
这一步是为了保证体系的稳定,否则在执行后面的操作时,体系会发生崩溃。
数据产出
前几个步骤都是准备工作,为的就是最后的数据产出这一步。数据产出的这一过程就是之前提到的基本原理中力场和势能的应用,要在整个系统平衡后进行。该过程可记录下体系中粒子随时间的变化的坐标、速度和能量。为了可观测到研究体系的性质,并保证此过程具有可重复性,模拟的时间(步长)一定要够久。
当完成以上步骤后,就可以对体系的轨迹进行分析处理,观察体系具有什么现象或性质。
总结
随着算力不断地提升,计算小体系下的毫秒级别和百万原子体系已经可以实现。而且MD模拟也衍生出来更多分支,如增强采样的RMED;适用于大分子体系的粗粒化模拟;提升计算速度的DMD等。MD模拟的参数是由经验参数组成的;模拟可以提供详细的构象分布和时间序列,是对实验的一种补充。所以说这是实验支撑理论,理论补充实验的一个过程。当然经验势函数也是有局限性的,针对不同体系的准确性会有差异。为了提升势函数的精确性及计算效率性,研究人员也在不断改进完善算法。
希望该文能让初学者对分子动力学模拟有初步的了解,欢迎大家一起讨论CADD的相关内容!
GROMACS教程网址 http://www.mdtutorials.com/gmx/
参考文献
[1]. Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for All[J]. Neuron. 2018 Sep 19;99(6):1129-1143.
[2]. 李文飞, 张建, 王骏, 王炜. 生物大分子多尺度理论和计算方法[J]. 物理学报, 2015, 64(9): 098701.
[3]. Wang Y, Liu T, Xie J, Cheng M, Sun L, Zhang S, Xin J, Zhang N. A review on application of molecular simulation technology in food molecules interaction[J]. Curr Res Food Sci. 2022 Oct 11;5:1873-1881.
[4]. 王存新. 蛋白质模拟:原理,发展和应用[M]. 科学出版社, 2016.
[5]. 刘冠辰.分子动力学模拟及在生物大分子模拟领域的应用[J].吉林化工学院学报,2015,32(11):112-116.
[6]. 刘远超. 分子力场[EB/OL].https://blog.sciencenet.cn/blog-3330179-1197033.html.
[7]. Yunxiang Sun, Aleksandr Kakinen, Yanting Xing, Pouya Faridi, Aparna Nandakumar, Anthony W. Purcell, Thomas P. Davis, Pu Chun Ke and Feng Ding. Amyloid Self-Assembly of hIAPP8-20 via the Accumulation of Helical Oligomers, α-Helix to β-Sheet Transition, and Formation of β-Barrel Intermediates[J]. Small, 2019: e1805166.
[8]. Hess Berk, Kutzner Carsten, Spoel David Van Der, et al. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation [J]. Journal of chemical theory and computation, 2008,4(3): 435-447.
|
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2022-11-17 11:28
发表于 2022-11-17 11:28
 提升卡
提升卡


