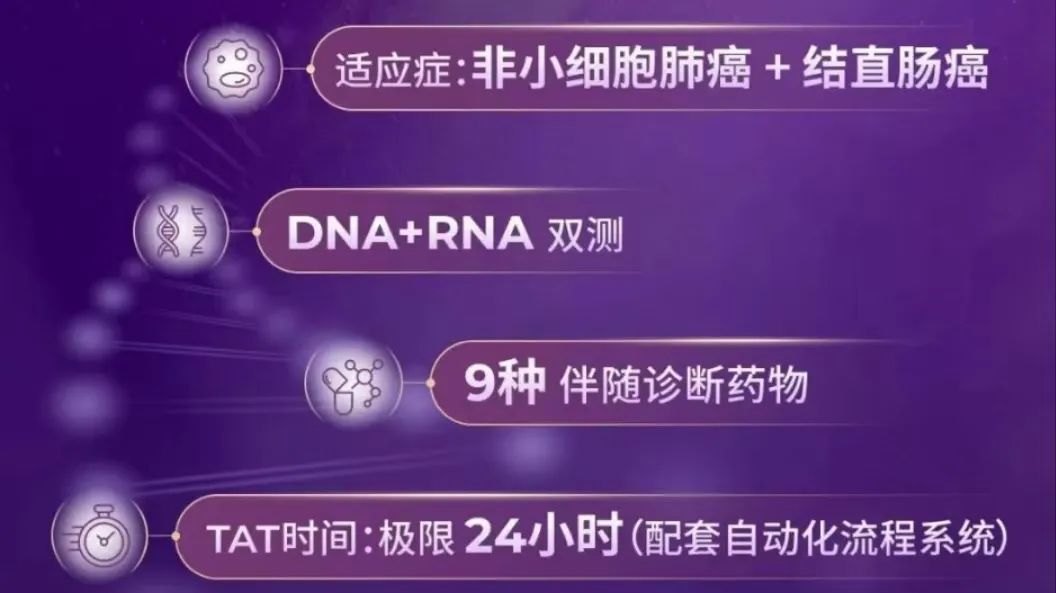
国内的CDx注册策略基石受到了巨大的挑战 昨天下午,药监局发布了新一批的医疗器械批准证明文件送达信息。 臻和喜提一张注册证:人类8基因突变联合检测试剂盒(可逆末端终止测序法) 本来,这没什么。 根据基因江湖九哥的统计,从2018年首证至今,已经有20张肿瘤NGS注册证获批,妥妥的从蓝海卷成了大红海。 “不过是又多了一张而已” 但如果细看这张注册证,它可能会改变的,不仅仅是臻和这一家公司。 在我的认知里,它在行业的原有规则之外,我看不懂。 老规矩,三部分,从小往大聊: 基本信息(这是小食) 可能的优势(这是前菜) 争议在哪(这是主菜)
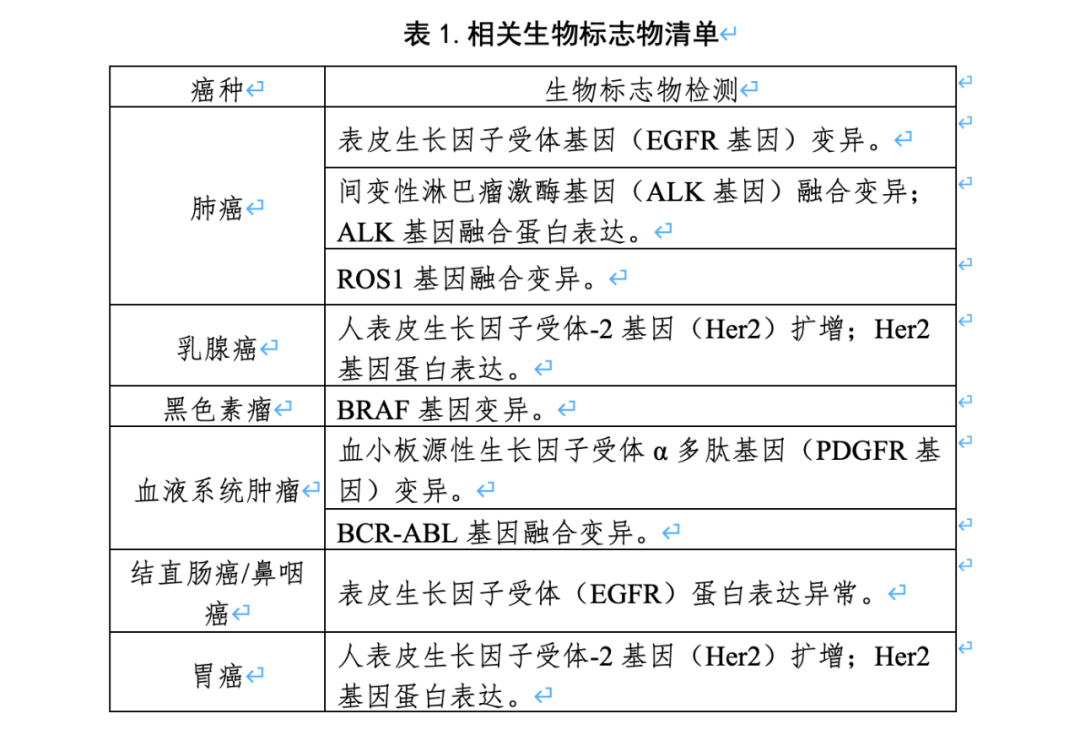
PS:这篇仅代表个人观点。如果我俩理解不一样,那么你是对的。欢迎留言区探讨(今天文末真有东西)。 臻和的动作是快的,第一时间就发布了官方海报,列出了四大优势: 我本来想找D总要一份说明书,看看这四大优势的细节。D总听闻非常感动,然后拒绝了我 ——以下由人工不智能小模型DeepDream生成——EGFR/ALK/ROS1/KRAS/PIK3CA/BRAF/ERBB2/METEGFR(Ex19Del、L858R):吉非替尼、埃克替尼、阿法替尼、厄洛替尼EGFR(Ex19Del、L858R、T790M):奥希替尼ALK重排(融合)、ROS1重排(融合):克唑替尼扩增子测序,DNA+RNA,其中RNA主要负责测融合突变。结合上面的信息,这张注册证对于臻和的价值可能不止“多了一张证”。 RNA在融合检测上相比于DNA的优势毋庸置疑,但更大的优势可能体现在“收费”的上限上。众所周知,在厮杀成红海的肿瘤NGS市场,进院价=收费价*折扣。以北京为例,肿瘤组织脱氧核糖核苷酸(DNA)测序的价格被限定在3800/例。对于单DNA测序的肿瘤NGS产品来说,这个天花板并不高,基因数一多就不容易算过来账。我们的朋友,公众号“小李叨叨”的叨哥在谈及这张注册证时表示:DNA+RNA双测的优势就在于它还可以组合另一个编码。还是以北京为例,通过组合“组织/细胞荧光定量脱氧核糖核酸(RNA)多聚酶链式反应检查诊断”这个项目,可以在3800/例的DNA天花板基础上扩展出640+500/对引物的价格上限。这张注册证给予了臻和在入院市场相比竞品更为灵活的价格策略。相比于更为主流的杂交捕获路线,扩增子的优势在于速度快、成本低。当然凡事有得必有舍,选择扩增子法的局限性也很明显。扩增子法对cfDNA和FFPE样本的检测需要较高的技术门槛,国内外应用的并不多,且对于未知突变引物设计也存在一定的难度。而且多重不容易做大,panel大了,对于扩增子引物之间的平衡需要做不少优化。这也许可以解释为什么同技术路线的“臻耀安”只做了200多个基因的panel。对感染领域tNGS有了解的朋友可以简单的类比多重PCR tNGS和探针捕获tNGS的优劣势就行。——这样一个“一部分管肺癌、一部分管肠癌”的产品,似乎突破了IVD注册中对于“单一注册单元”的限制。但其实这没什么,时间窗口的问题,不是今天的重点,暂且按下不表。这个试剂盒所有的伴随诊断都是采用与原研伴随诊断试剂一致性比对的方式验证其伴随诊断用途。与原研试剂比对这条路径没什么疑问,疑问出在MET这个基因上。在MET基因外显子14跳跃突变上,臻和比对的是艾德的AmoyDx® Pan Lung Cancer PCR Panel。在《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》中,给出过可以通过“一致性比对”来获批伴随诊断预期用途的基因列表:而后在2024年发布的《基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验注册审查指导原则》中,对于非小细胞肺癌增加了BRAF V600E这个位点。也就是说,至今为止没有任何公开文件提及MET 14外显子跳跃突变可以通过“一致性比对”来验证其伴随诊断用途。即使在“非原研伴随诊断注册审查指导原则”中预留了“该清单会随着科学认知的深入及相关产品的临床应用情况适时更新”。但不管从哪个角度看,MET 14外显子跳跃突变似乎都不满足被“增补”进清单的条件。如果MET可以,那是不是别的不在列表里的基因也可以?我们都希望“站在巨人的肩膀上”,但似乎得有个规则?这可能才是这张注册证给国内肿瘤NGS市场投下的那枚“炸弹”。原研伴随诊断的开发不管是从时间成本还是经济成本上,都远超非原研。而大家之所依然热衷去拿药企的原研伴随诊断合作,是因为随着国内市场竞争的不断规范化,“独占”的原研伴随诊断能够为获批试剂盒创造一个有利的竞争时间窗口。而这次的MET,给这个“时间窗口”的优势,打下了一个问号。原研的AmoyDx® Pan Lung Cancer PCR Panel在国内的获批时间是2024年9月。换句话说,如果原研伴随诊断的领先时间窗口如此短暂,这是否依然能成为肿瘤基因检测的核心竞争壁垒之一?如果它不是,那么大家是否依然还有动力去做原研伴随诊断?“为啥我要花几年时间和药企合作,成果是支持竞品做一致性比对???”更为好玩的是,这个“非原研”可能比“原研”的获批更“有优势”。Amoy的试剂盒在国内获批的时候是“附条件批准”。而可靠的消息源表示,臻和的MET伴随诊断没有附条件。在2024年8月的《中华人民共和国医疗器械管理法(草案征求意见稿)》中,给到了医疗器械注册证转让的想象空间。当时我们认为这会极大的提升创新的动力——拿证、拿新证、拿独占证。但如果原研伴随诊断可以被fast follow,它的交易价值可能就要大打折扣了。需要注意的是,从临床试验的时间推断,臻和选择Amoy这个试剂盒做一致性对比的时间,是早于Amoy试剂盒获批的,这也就意味着fast follow的时间间隔,会被压缩的很短。——这里其实还有个疑问,按照时间推断,比对的试剂盒应该是日本或者欧洲的版本,而Amoy在日本获批“特泊替尼”伴随诊断时,是从采用方法学比对Archer获批,并未参与默克药物临床,这似乎并不属于原研伴随诊断。深究细节的话,这种和“非原研”进行比对然后获批,是真的成立吗?如果回报显著降低,大家当“第一人”的意愿也一定会降低。“什么基因可以通过一致性比对获批伴随诊断预期用途”,将会是这张注册证留下的会被一直讨论的话题。如果,我是说如果,注册证不再是那么显性的竞争壁垒。
| 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号