它只管提要求,办法还得自己想......
在分析Natera Q3财报的时候,我们提了一句话
那么,当前在监管机构的认知里认可哪些干预的场景? 这个问题一直没有一个明确的说法。 恰逢年末,它来了,带着自己的“小作文”向我们走来了。
FDA的这个“指引”比较系统的阐述了监管机构当前对于MRD和治疗决策之间的认知、理解、思考。 阅读前提示:
带着这样的认知,再来看这篇指引,就会清晰很多:
PS:这篇所有引用材料均来自公开信息,仅代表个人观点。如果我俩理解不一样,那么你是对的。“指引”全文扫描二维码自行下载
01 “治疗升降级” 在FDA看来,MRD可作为药物临床试验入组时的生物标志物,用于富集/识别“早期治疗(包括手术、放疗、新辅助治疗、辅助治疗等)后的高风险患者”。
而富集的目的是进行治疗干预,FDA支持的探索是:
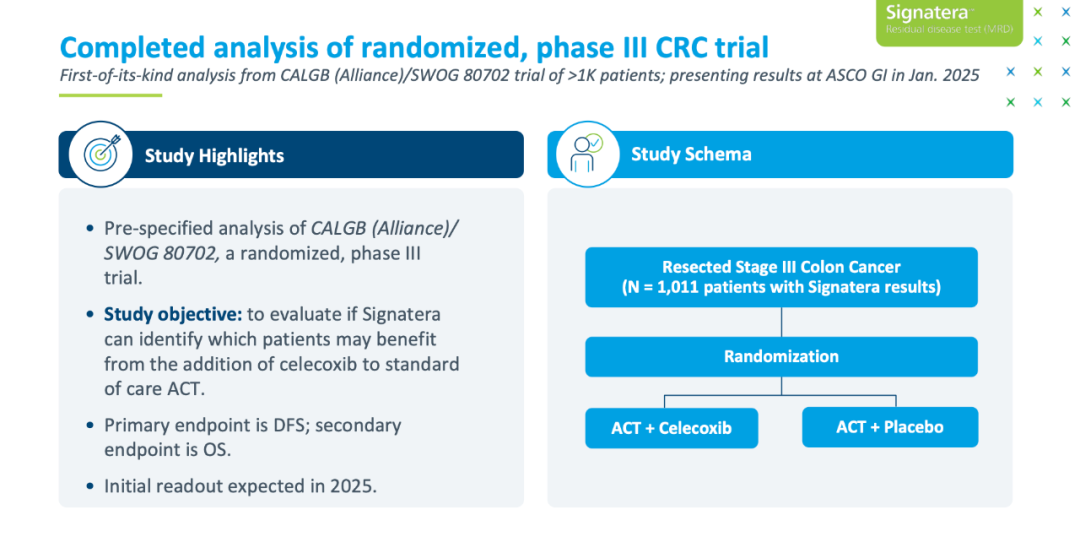
意思是再SOC的基础上增加某种疗法。 比如像CALGB/SWOG 80702那样增加Celecoxib
治疗降级可以是一个毒性更小的新方案,也可以是对现有SOC的“疗程/剂量”进行缩减(包括取消治疗)。 比如像下图中的VEGA就是一个典型的降级研究
场景的饼画完了,下面是要求: 于治疗升级而言,FDA认为高特异性和PPV是关键性能。 于治疗降级而言,FDA认为高敏感性和NPV是关键性能。 ——都是宁漏勿错,突出一个底线思维:不要带来额外伤害。
而不管是升级还是降级,临床试验的门槛都不低。 临床试验的主要终点应该是DFS(辅助治疗)、EFS(新辅助治疗)或者OS。 注意上面的“或者”,就算不把OS列为主要终点,也需要证明OS没有受到负面影响。
想要在临床试验早期就出个中期分析提振下士气? FDA表示“哒咩”: 不建议进行早期的中期分析,中期分析需要在大多数患者已经完成治疗后再进行。
这两重要求下,带来的就是临床试验的时间会被拉长。 大队列+3-5年的长时间研究,怕是省不了了。 燃烧的,都是经费。
02 “临床试验的终点” 比较基础的操作是用于“药物抗肿瘤活性”的评估,包括确定最佳剂量等。 更大的潜在价值在于作为临床试验的早期终点。
在FDA看来,MRD的潜力在于: 对于(新)辅助治疗的疗效评估,ctDNA变化或者清零(MRD阳转阴)可能优于传统的基于影像学的评估策略(比如ORR)。 但要求依然是不低的:
对于第一个吃螃蟹的人来说,难度不小。 当然,有些问题证明第一次很难,往后则容易不少。 谁先吃个螃蟹?
03 “路线选择” Tumor-inform一定好于Tumor-agnostic吗? 至少在FDA看来,未必。 在FDA的认知里,Tumor-inform和Tumor-agnostic各有各的好
优点:因为是基于肿瘤组织测序来构建的MRD panel,所以相对来说特异性可能会更高(结合前面的场景,这也就意味可能更贴合FDA对于“治疗升级”的性能要求)。 局限性:TAT时间会长,无法检测到“新发突变”,能通过组织测序筛选出的靶点数也影响了其后续性能。 基于这个局限性,FDA认为: Tumor-inform路线的MRD在不同特征的肿瘤中会有显著的性能差异,比如在高TMB(肿瘤突变负荷)且突变特征明确的肿瘤中会更可靠。
优点:TAT时间短。 局限性:“漏检”的风险比tumor-inform要大,毕竟肿瘤突变有着非常明显的长尾效应,具体到个体上的差异更显著。 不过对于这个局限性,FDA也给出了“一个思路” ——上WGS,突出一个“应有尽有”,还有多组学的加持。
当然这就显得labcorp有点像个憨批…… 独树一帜的tumor-inform WGS MRD路线,在FDA的视角下多少有点脱了裤子放屁?
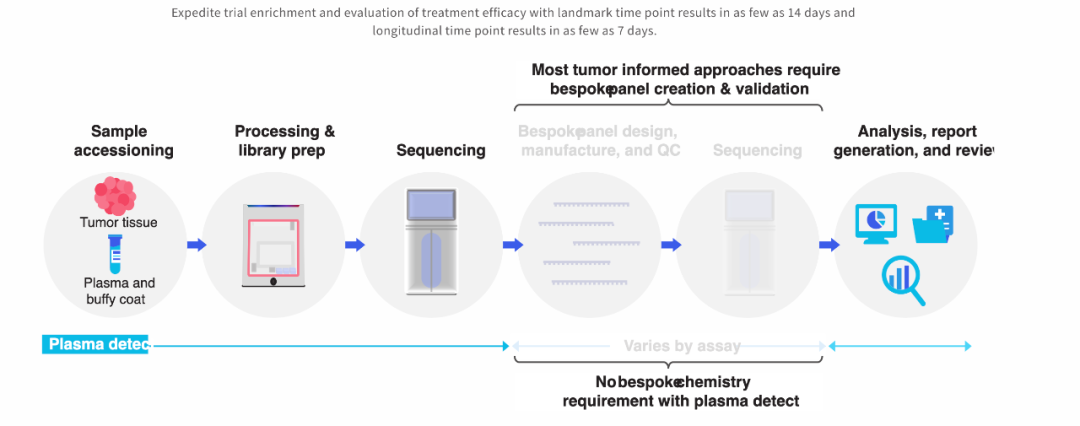
04 “分析性能验证” tumor-inform+定制化panel该如何做分析性能验证? FDA这一次给出了相对容易的解法。 “取合集来验证即可” ——虽然怎么证明“这是一个能代表人群特征的合集”依然是个难题,但总比之前不知道怎么验证要好。
FDA同时提到了参考品的事情 ——如何有一套参考品可以横向比较不同策略的MRD产品?
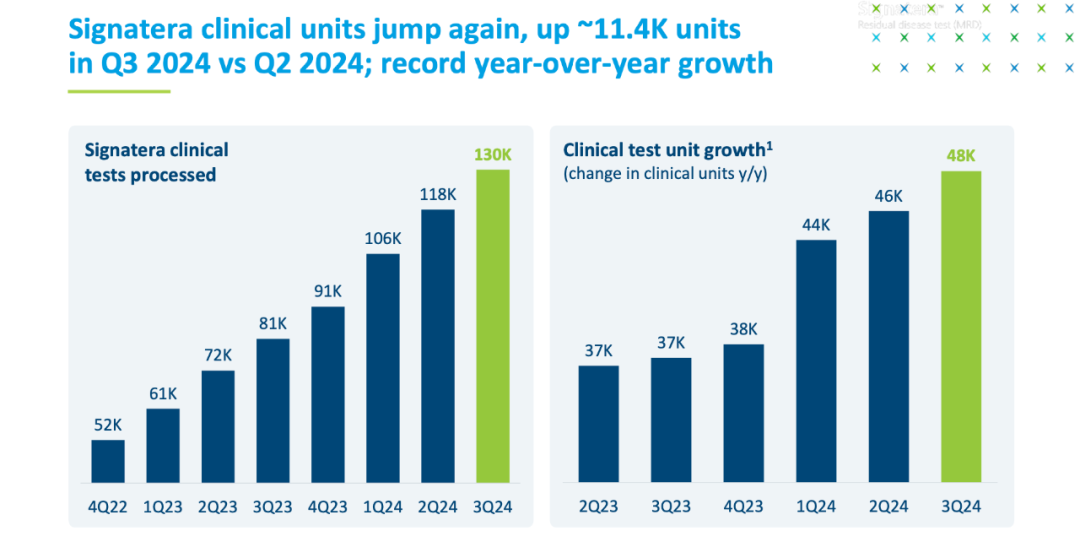
05 “出发吧,勇士” 对于在肿瘤NGS里艰难前行的各位,MRD绝对是一片沙漠绿洲。 尤其是Natera把检测量带到每个月10万+以后
虽然这个指引不能作为“指南”,但基本代表了当前监管机构对于MRD的“接纳边界”。 饼,是画的很大的。 不管是“临床试验终点”对于药企合作业务的促进,还是“治疗升降级决策”对于临床应用的刺激,单拎出来任何一个都是能让行业再创辉煌的存在。 挑战也是存在的。 如何去证明上面的“机会”不是一个伪命题? FDA给出了指引算是为大家标出了绿洲的坐标。 那么,剩下的问题就是: 谁,能成为头号玩家?
|


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号