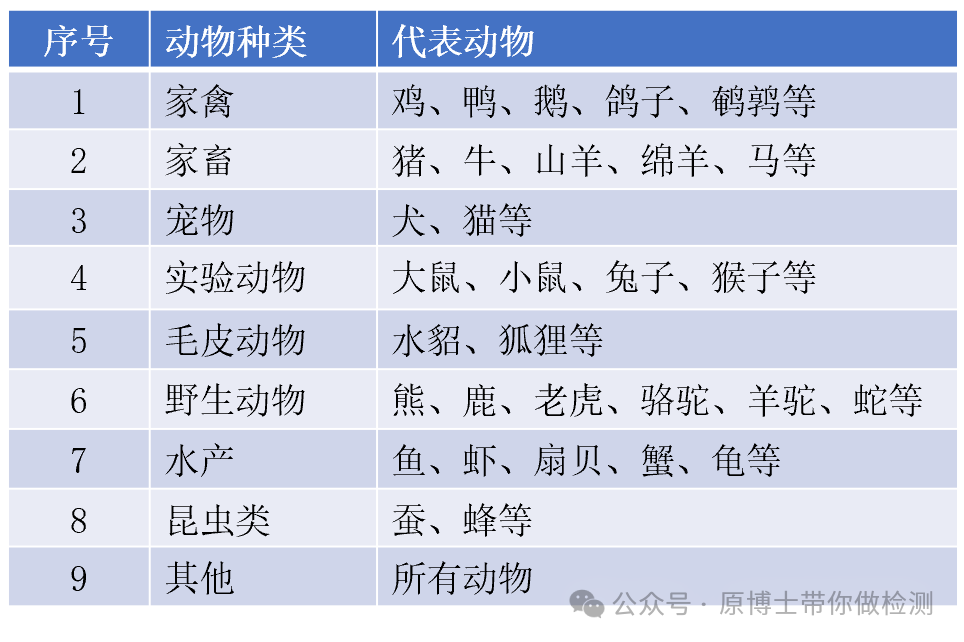
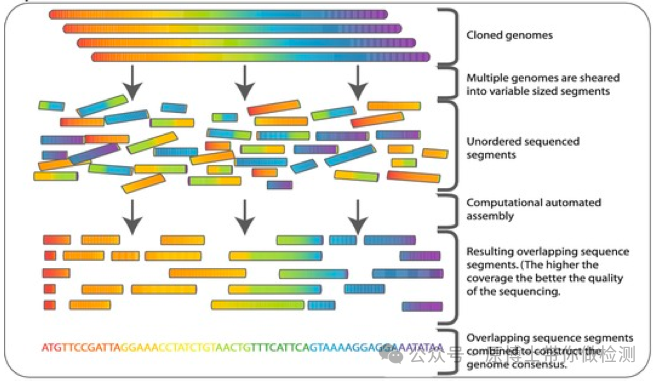
2024年9月20日,由中国兽医协会兽医实验室检测分会牵头起草的《动物病原微生物宏基因组高通量测序技术规范专家共识(2024版)》,正式发布。该专家共识为国内兽医领域首个宏基因组测序相关的指导性文件,为高通量测序技术在兽医领域规范使用奠定了基础。
[摘要] 宏基因组高通量测序技术作为一项新兴的、通用的微生物检测工具,可以识别所有微生物,在动物病原微生物临床检测中发挥了重要作用,被广泛应用于动物多病原检测、未知病原诊断、疫病流行病学调查、病原溯源等多种场景。但在兽医检测行业,宏基因组高通量测序技术尚未形成一致性、规范性认识。本文从实验室建设及管理、实验操作、生物信息学分析、结果判定、质量控制、报告审核等方面阐述了宏基因组高通量测序技术在动物病原微生物临床检测中应用的专家共识,并提出规范性要求和建议。 [关键词] 宏基因组高通量测序技术;动物病原微生物;专家共识 过去几十年来,禽流感、非洲猪瘟、口蹄疫等动物传染病给畜牧业造成了重大损失,同时动物来源的传染病病原体对食品安全和公共卫生健康构成了严重的威胁。据世界卫生组织报告,全球新发传染病中约有60%的传染病和75%的新兴人类病原体来自动物。有效监测、控制动物病原微生物的发生对维护人类、动物和生态系统“同一个健康”至关重要。
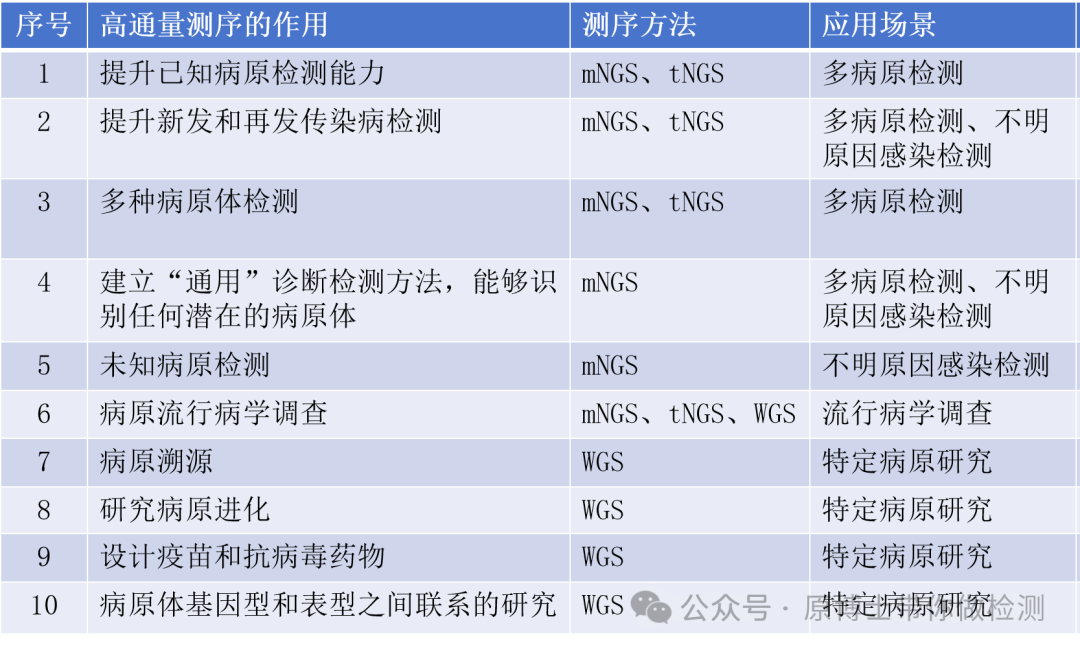
微生物高通量测序(High-throughput Sequencing, HTS)方法主要包括全基因组测序(Whole genome sequencing,WGS)、宏基因组测序(metagenomic next–generation sequencing,mNGS)、靶向测序(Targeted Next-generation Sequencing,tNGS)。
.(A) WGS of a pure organism from cultured growth. (B) tNGS directly from specimen. (C) mNGS from clinical specimens. 其中,宏基因组高通量测序技术不依赖于传统的微生物培养,无需特异性扩增,直接对临床样品中的核酸进行高通量测序,通过与数据库进行比对分析,根据比对到的序列信息来判断样品包含的病原微生物种类,能够快速、客观地检测临床样品中的所有微生物。 宏基因组高通量测序技术可用于动物及动物产品(动物源性食品、疫苗、饲料、生物材料、培养物或分离物等)中的微生物(包括病毒、细菌、真菌、寄生虫、放线菌、衣原体、支原体、立克次体、螺旋体)的测序,适用于二代和三代测序平台。 基于临床的宏基因组测序是对临床或环境样本的所有基因组(DNA 和/或 RNA)进行测序。 一些科研服务类测序公司将宏基因组测序限定为仅用于DNA测序,将宏转录组测序定义为RNA测序,不符合医学和兽医临床对宏基因测序的定义。 病原微生物是指可以侵犯宿主,引起感染甚至传染病的微生物,或称病原体。病原微生物包括细菌、真菌、病毒、螺旋体、支原体、立克次体、衣原体、寄生虫(原虫、蠕虫、医学昆虫)等。
世界动物卫生组织(World Organization for Animal Health,WOAH)陆生动物诊断测试和疫苗手册推荐了高通量测序技术在动物病原微生物检测上的应用场景及检测目的:
现在我国尚未出台动物病原微生物宏基因组高通量测序技术应用的规范性标准,国内部分兽医实验室建立了各自的宏基因组高通量测序平台,但还没有形成行业内规范性、系统性、一致性的认知。为了满足兽医临床测序需要,本共识将对如何开展动物病原微生物宏基因组高通量测序提出具体的指导性意见。 1.1生物安全要求 宏基因组高通量测序的动物种类和病原微生物种类范围广泛,可能存在高致病性病原微生物,开展动物病原微生物宏基因组高通量测序的实验室应具备生物安全二级(biosafety level 2,BSL2)及以上的条件和相关资质,并严格按照农业农村部要求及相关法律法规进行实验操作。
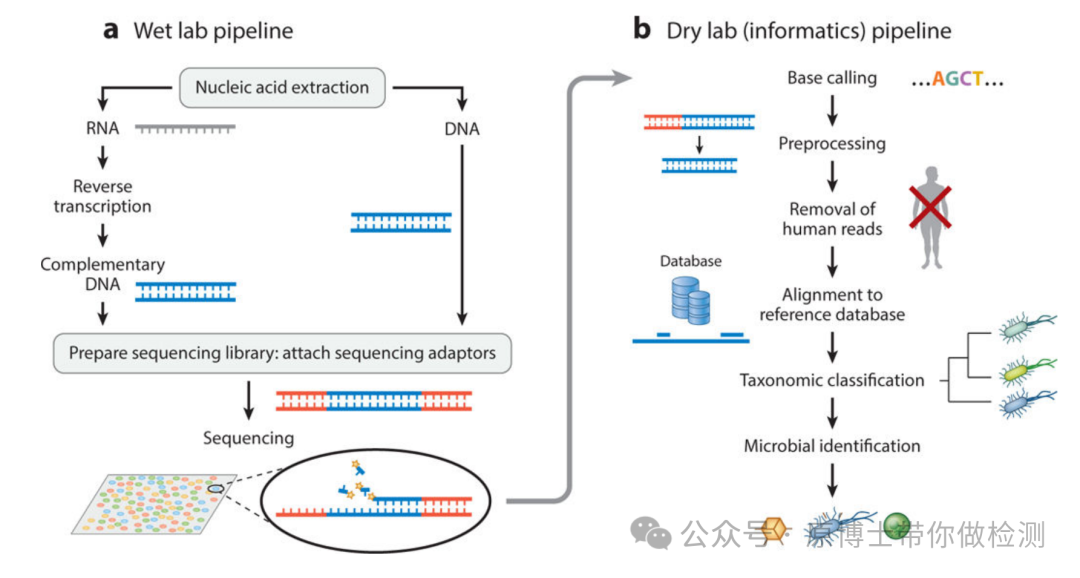
1.2 实验室结构布局 宏基因组高通量测序整个流程通常分为湿实验和干实验两部分。 湿实验流程是指从样品处理到测序数据产生的整个过程,需要在标准的实验室完成,一般包括:样品前处理、核酸提取、文库制备、测序; 干实验流程是指对测序数据进行生物信息学分析及结果审核和出具报告的过程。
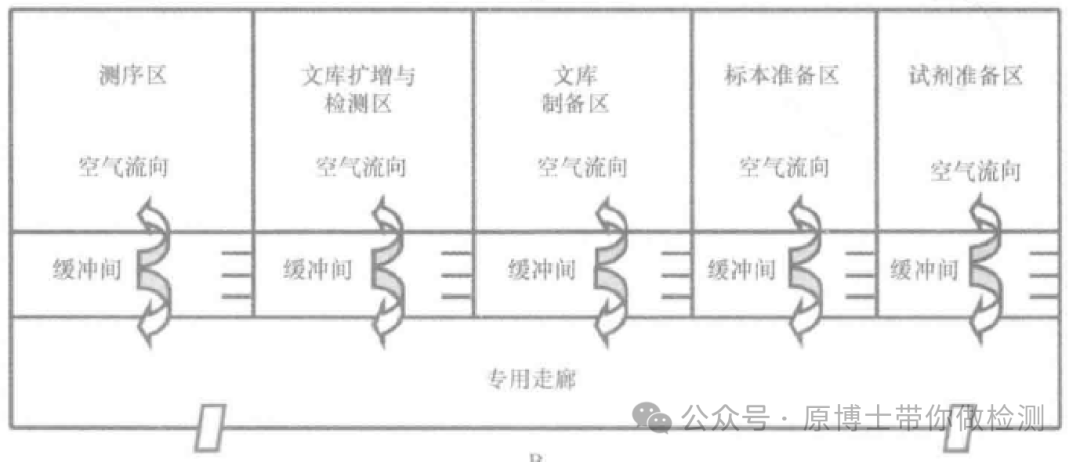
宏基因组高通量测序湿实验的每一个环节都要严格控制气溶胶污染。 实验室具备的基本功能区域应包括:体系配制室、样品处理室、核酸提取室、文库制备室、测序室,各实验室应配备能满足实验需求和质量控制要求的实验设备,人员、试剂和耗材应单向流动。 测序的气溶胶污染风险不比荧光定量PCR低,由于环节更多,检测病原更多,污染的风险其实更高。气溶胶污染控制不好会导至越来越多的假阳性出现。 测序实验室的分区:体系配制室、样品处理室、核酸提取室、文库制备室、测序室。是对一个测序实验室的最低要求,如果涉及靶向扩增,文库制备室还需增加一个PCR扩增的房间。
1.3 人员配置 实验室要求相关工作人员必须具有相应专业背景和专业技能,包括: 湿实验要求实验人员应具备分子生物学专业技能,保证实验规范性操作; 数据信息分析人员应具备生物信息学专业技能; 结果和报告审核人员应具备动物医学专业背景和兽医临床经验。 所有人员均须通过岗位相关的培训和考核方能上岗。 1.4 测序平台 高通量测序技术包括第二代测序技术(next generation sequencing technique,NGS)和第三代测序技术(third generation sequencing technique,TGS)。 NGS以DNA纳米球测序和边合成边测序技术为主,测序数据量大,准确性高,测序成本低,但测序读长较短,拼接复杂、长基因组时较困难;
TGS以纳米孔测序和单分子荧光信号测序两种技术为主,测序读长长,可拼接长基因组序列,但数据量较低,准确性较低、测序成本较高,可实时产出数据,设备可便携。
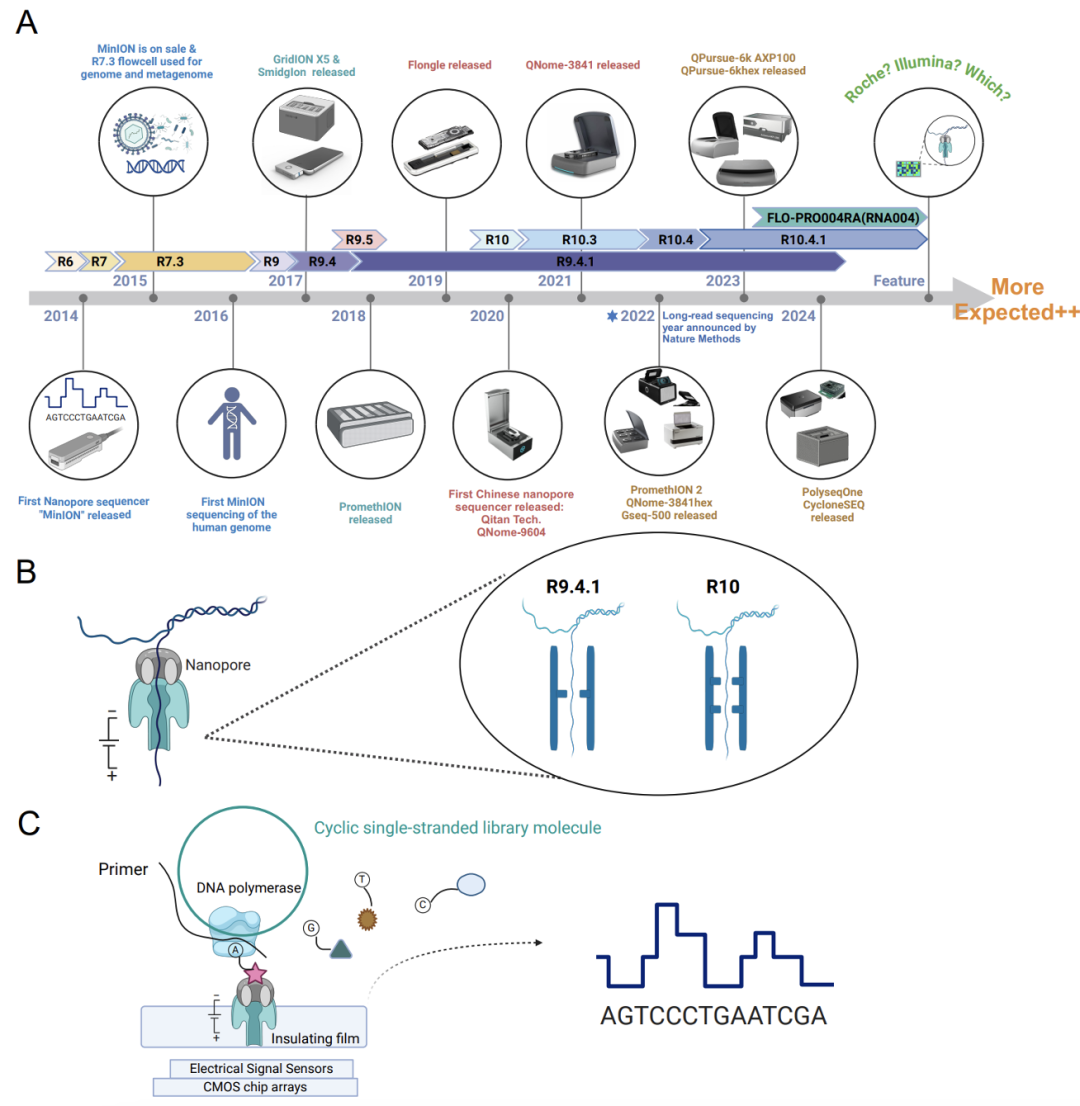
过去十年纳米孔测序的进展 文献14 A: 过去十年各种纳米孔测序平台和试剂盒的开发和发布时间。关键里程碑包括MinION、GridION、Flongle、PromethION和各种中国纳米孔平台的发布;B: 纳米孔测序原理示意图;C: 中国纳米孔测序仪Gseq-500和AXP-100的测序原理,其采用了纳米孔合成测序(NSBS)策略,在DNA聚合酶驱动的DNA链合成过程中,在碱基切割后标签进行测序,以确定核苷酸序列。 各实验室可根据自身测序样品数量、测序长度、准确度、数据量、时效性和测序成本等因素选择适合的测序方法和测序平台。 测序平台没有好坏,只是各自特点不同,应用场景不同。关于测序平台的几个误区: 1.测序准确性:指的是单条序列的准确性,二代测序可以达到99.9%,三代测序90-99%。很多公司说的是后期通过算法根据多条序列的结果进行修正的准确性,那就不是仪器的性能了。 2.测序长度:二代测序的长度比较确定,设定好读长就确定了,比如50bp、100bp,150bp;三代测序的长度不容易确定,尤其是做临床样品的宏基因组测序时,通常N50能到1k就很好了,长读长通常是对纯细菌或物种基因组的测序获得的。 3.测序序列数与数据量:二代测序比较好算,序列数×长度=数据量,比如10M×100bp=1G数据量;三代测序宏基因组测序就比较难以计算,如果以获得1G数据量,按照平均1k的长度,则序列数为1M。 二代测序宏基因组测序的最低要求是序列数为10M/样品,如果换算到三代测序上需要的数据量是10G,就已经超过很多三代测序的单张芯片产出了。 4.RNA直接测序:针对的是纯化过的RNA,而非RNA病毒培养物或临床样品中的RNA。 小结: 二代测序由于测序数据量更大,单位数据量成本更低,结果更准确,适合于宏基因组测序和多病原靶向测序; 三代测序由于数据量相对较小、单位数据量成本较高,读长更长,更适合于全基因组测序。对时效要求很高情况下的宏基因组测序和多病原靶向测序。 如果问我不同测序平台选择的意见,我的意见是:二代和三代测序完全是互补的关系,因此,有条件的实验室二代三代测序仪都需要!
文献13 2.1 样品采集及制备
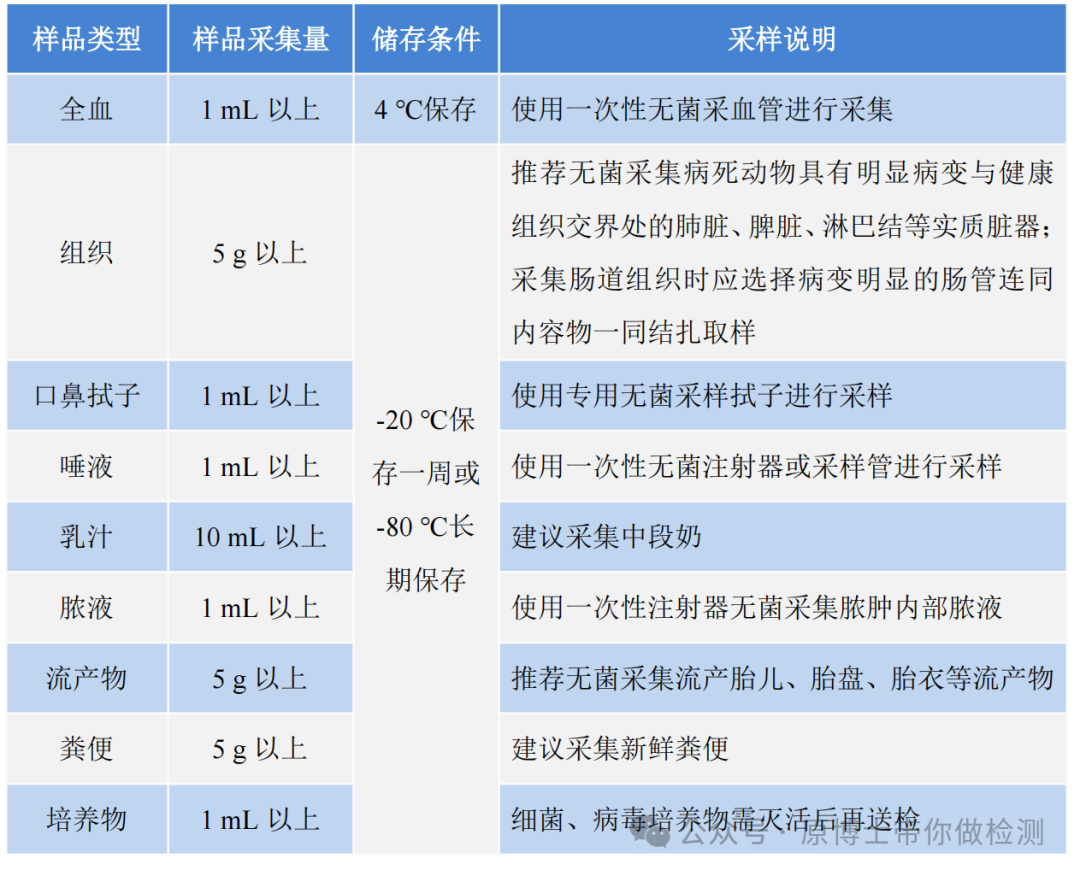
样品质量和适用性直接影响动物疫病的宏基因组高通量测序实验结果。因此,样品采集应尽量选择感染部位的体液或新鲜组织,以提高测序的灵敏度。采集过程中应执行无菌操作,避免样品被污染。样品采集过程中应使用无菌采样管、采样袋等无菌采样,每个样品须单独保存,在包装外标记清楚样品编号、样品名称、物种来源、采样日期、采样地点、送检人名称及联系方式、送检单位、病例信息及测序目的等信息。 表1 部分样品采集注意事项表
样品采集后应以最快最直接的方式送往实验室,运输过程中需使用低温容器,以免发生核酸降解,并防止样品泄漏和污染。若无法及时送检,应按规定条件进行保存。 在动物疫病领域,样品类型复杂,不同样品类型需要采用不同的前处理方式和核酸提取方法。 样品制备时应遵守从洁净到非洁净、从简单到复杂的规则,优先制备全血、组织等相对洁净的样品,再制备口鼻拭子、粪便等非洁净的样品。 兽医领域样品的采集和制备是测序能否成功的关键因素: 1.采样:需结合临床,采集靶器官病变与健康组织交界处,新鲜样品、无菌采样。 2.运输:建议尽快送检,-20 ℃保存一周或在-80 ℃长期保存。每个样品需要独立包装。 3.制样:先从洁净的实质性脏器和血液,到呼吸道样品,再到肠道、粪便、环境样品。 4.合样:宏基因组测序方法的灵敏度本来就不高,不建议合样检测。 2.2 核酸提取 提取样品核酸时,可选用自动化提取或者手动提取。根据测序目的和建库要求,提取方法可选用DNA提取、RNA提取或者DNA和RNA共提取。宏基因组高通量测序样品类型复杂,对文库核酸的纯度和浓度要求高,且细菌、真菌、寄生虫、支原体等微生物提取难度大,建议使用经过验证可用于宏基因组高通量测序的核酸提取试剂。核酸质量应符合建库要求。 测序样品的核酸提取比荧光定量PCR的提取要求高,需要保证提取的纯度、浓度和无污染。建议采用经过验证可用于测序的提取试剂。 对于粪便和寄生虫等特殊样品,需要采用专用的提取试剂。
2.3 文库制备 文库制备是指将基因组DNA通过打断、连接测序接头等方式形成适用于测序仪器读取的形式。根据测序目的和样品类型选择合适的测序平台和建库方法,文库制备方法可分别选用DNA文库制备、RNA文库制备、DNA和RNA共建库。实验室在建库过程中需要对核心技术环节质量进行评估,包括核酸的投入量、单/双标签、片段大小、文库的浓度和纯度等,确认文库质量符合仪器测序模式和数据质量要求。 DNA和RNA共建库更适合于宏基因组测序对未知病原的检测。 很多机构用宏转录组的建库流程代替RNA病毒的建库流程,两者有相识的地方也有区别,理论上,宏转录组应在提取过程中去掉RNA病毒核酸,只针对mRNA建库,而RNA病毒建库应去除tRNA、mRNA等干扰,只针对RNA病毒核酸建库。
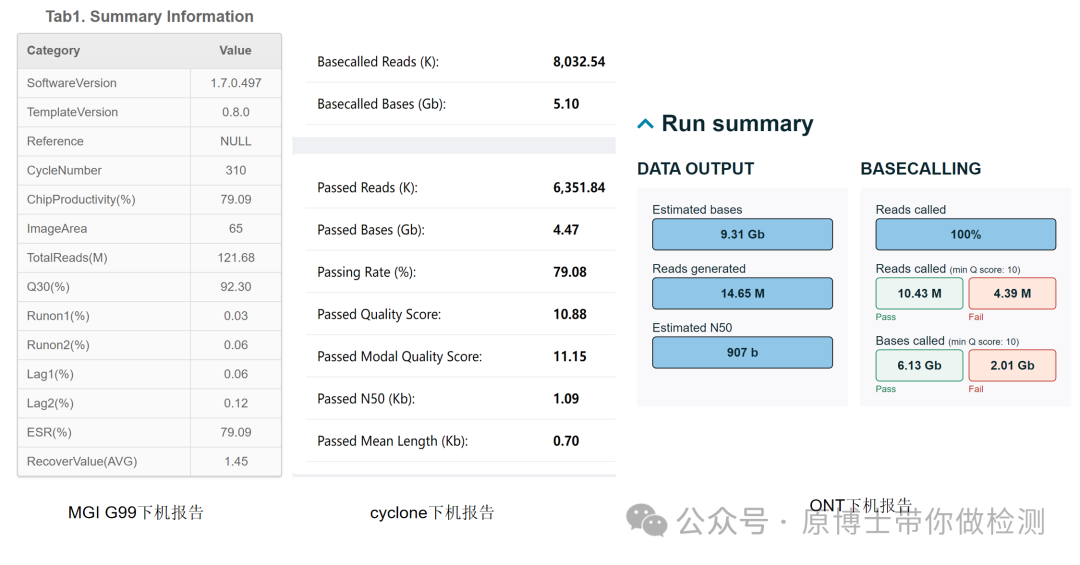
文献13 2.4 上机测序 测序仪与配套试剂、耗材通常为封闭一体化设计,实验室按照选用的测序仪器说明书进行操作,并根据不同型号仪器对文库进行前处理。测序完成后,需确认碱基识别质量、N50、文库有效数据量、数据拆分率等数值符合测序要求。
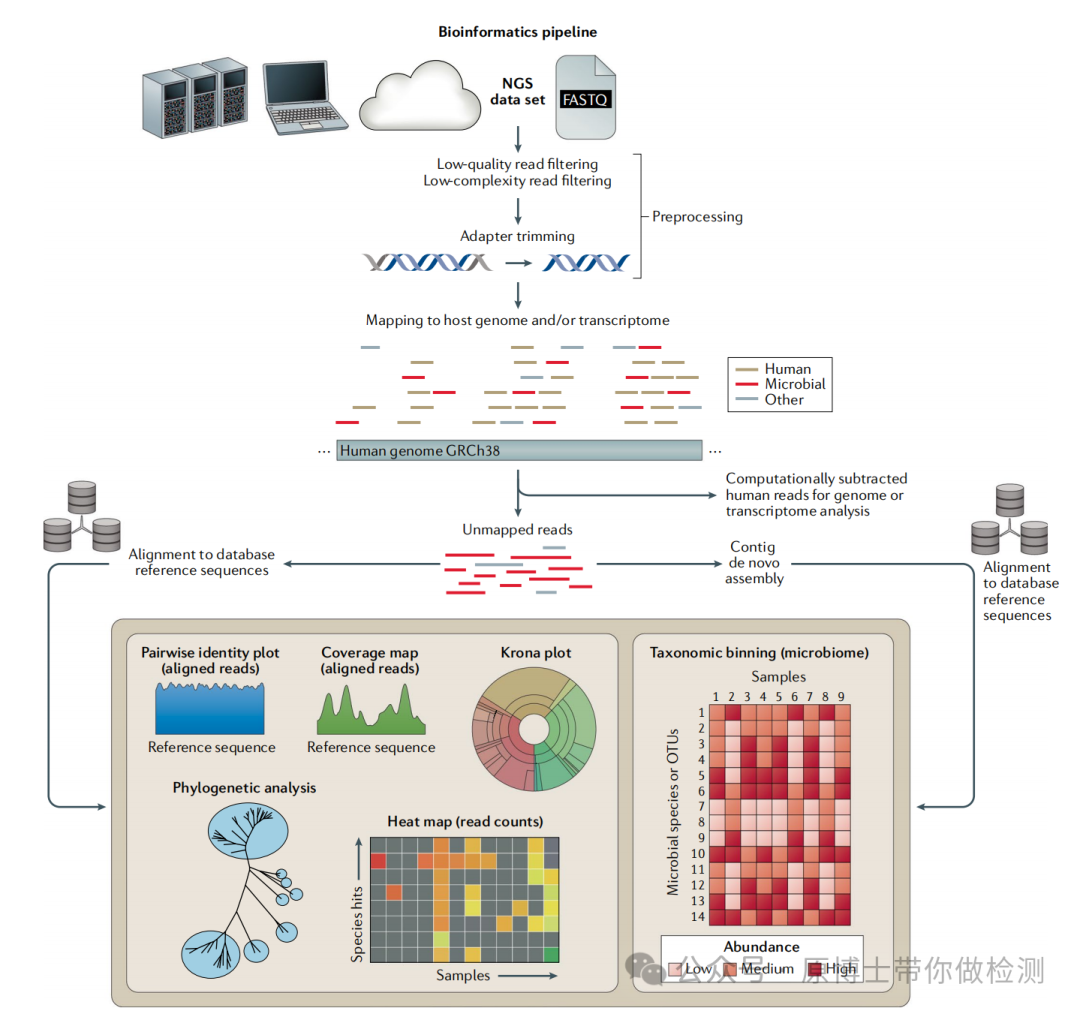
03 生物信息分析是对测到的原始序列进行数据分析和处理,最终将测序数据转化为服务于临床兽医和生产人员的病原微生物报告的过程。
文献13
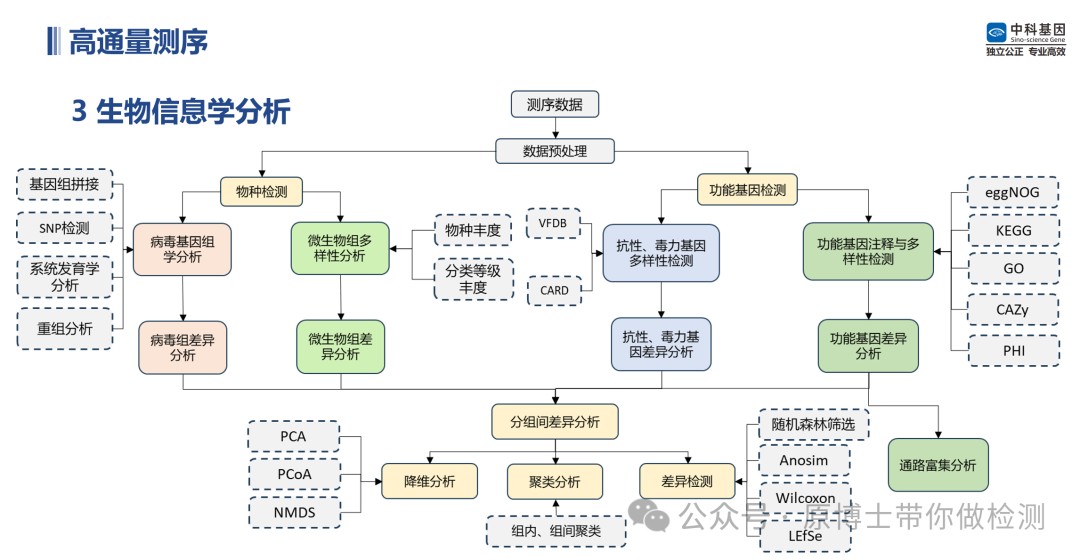
中科基因生物信息学分析流程 3.1 数据库 中国目前尚无专门用于动物病原微生物宏基因组高通量测序数据生物信息学分析的标准数据库,国内各测序实验室通常使用NCBI GeneBank、NCBI RefSeq、国家微生物科学数据中心发布的gcPathogen等公共数据库,但任何一个公共数据库都不能包含所有微生物基因组信息,且数据库中不可避免地存在一些注释错误或污染的序列导至的假阳性和新发病原体或病原发生变异导至的假阴性。 建议动物病原微生物宏基因组高通量测序实验室在公共数据库的基础上自建或使用商品化动物病原微生物数据库,该数据库应包含公共数据库信息、特定病原基因组信息,以及临床流行毒株基因组信息。
需要对动物病原微生物数据库进行定期更新、维护,及时补充微生物不同亚型或亚种以及新发病原体的基因组信息,提高数据比对的准确性和全面性。 实验室应建立专用的动物宿主数据库,该数据库应包含测序相关的宿主基因组信息,并定期更新、维护,及时补充测序所需宿主基因组的数据。
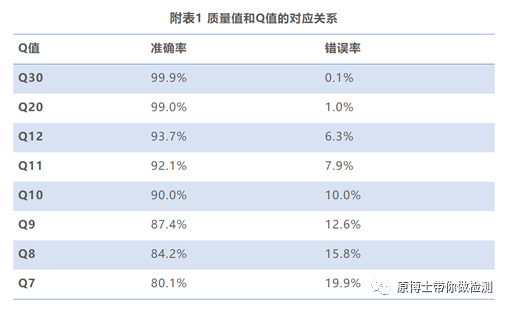
实验室需配备专用的高性能服务器,该服务器应可集成分析流程和数据库,对下机数据进行分析处理。 3. 2 数据分析 3.2.1 数据预处理 测序的原始数据是指样品测序获得的没有经过任何处理的全部测序数据,包括宿主序列、病原微生物序列、接头序列、标签序列,以FASTQ格式储存。原始数据去除接头序列、标签序列后得到的可用于比对的序列称为可用数据或纯净数据。 数据过滤指标建议:保留的有效读长序列不小于50 bp;二代高通量测序数据用Q30作为阈值去除低质量序列,三代高通量测序数据用Q7作为阈值去除低质量序列;应完全去除接头序列和重复序列。 常用软件有Trimmomatic、fastp、NanoPlot等。 碱基质量值(quality,Q): 体现测序过程中碱基识别的可信度和错误率,其计算方式为Q =-10lgP(P为碱基识别中的错误概率)。碱基 Q 值越大其识别错误的可能性越小,可信度就越高。 Q30:表示碱基识别错误的概率为 0.1%。 Q20:表示碱基识别错误的概率为1%。 Q10:表示碱基识别错误的概率为10%。 Q7:表示碱基识别错误的概率为19.9%。
Trimmomatic: 对 illumina 双端和单端数据执行各种有用的剪切任务。在命令行中提供了修剪步骤的选择及其关联参数。例如剪切特异性序列、低于阈值质量的序列等。 fastp:超快速的 FASTQ 预处理器,具有有用的质量控制和数据过滤功能。它可以通过一次扫描 FASTQ 数据来执行质量控制、适配器修剪、质量过滤、每次读取质量修剪和许多其他操作。 NanoPlot :用于长读长测序数据和比对的绘图工具
二代测序下机数据fastp质控图
三代测序下机数据NanoPlot质控图 3.2.2 去宿主 为了避免残留的宿主核酸对后续动物病原微生物分析产生影响,在动物病原微生物测序数据分析时,应根据宿主信息,去除测序数据中的宿主基因组数据。常用软件有BWA、Bowtie2、Minimap2等。 BWA: 是一个软件包,用于将 DNA 序列与大型参考基因组(例如人类基因组)进行映射。它由三种算法组成:BWA--backtrack、BWA-SW 和 BWA-MEM。第一种算法设计用于高达 100 bp 的 Illumina 序列读数,而其余两种算法用于较长的序列,范围从 70 bp 到几兆碱基。BWA-MEM 和 BWA-SW 具有相似的功能,例如支持长读长和嵌合比对,但通常推荐使用最新的 BWA-MEM,因为它更快、更准确。对于 70-100 bp Illumina 读长,BWA-MEM 的性能也优于 BWA-backtrack。 Bowtie2:是一种超快速且节省内存的工具,用于将测序读数与长参考序列进行比对。它特别擅长比对大约 50 到 100 或 1,000 个字符的读长,特别擅长比对相对较长的(例如哺乳动物)基因组。Bowtie 2 使用 FM 索引对基因组进行索引,以保持其内存占用较小。Bowtie 2 支持间隙对齐、局部对齐和双端对齐模式。 Minimap2 :是一种多功能序列比对程序,可将 DNA 或 mRNA 序列与大型参考数据库进行比对。典型用例包括:(1) 将 PacBio 或 Oxford Nanopore 基因组读数映射到人类基因组;(2) 发现长读长之间的重叠,错误率高达 ~15%;(3) PacBio Iso-Seq 或 Nanopore cDNA 或 Direct RNA reads 与参考基因组的剪接感知比对;(4) 对齐 Illumina 单端或双端读长;(5) 装配体到装配体对齐;(6) 两个密切相关物种之间的全基因组比对,差异低于 ~15%。
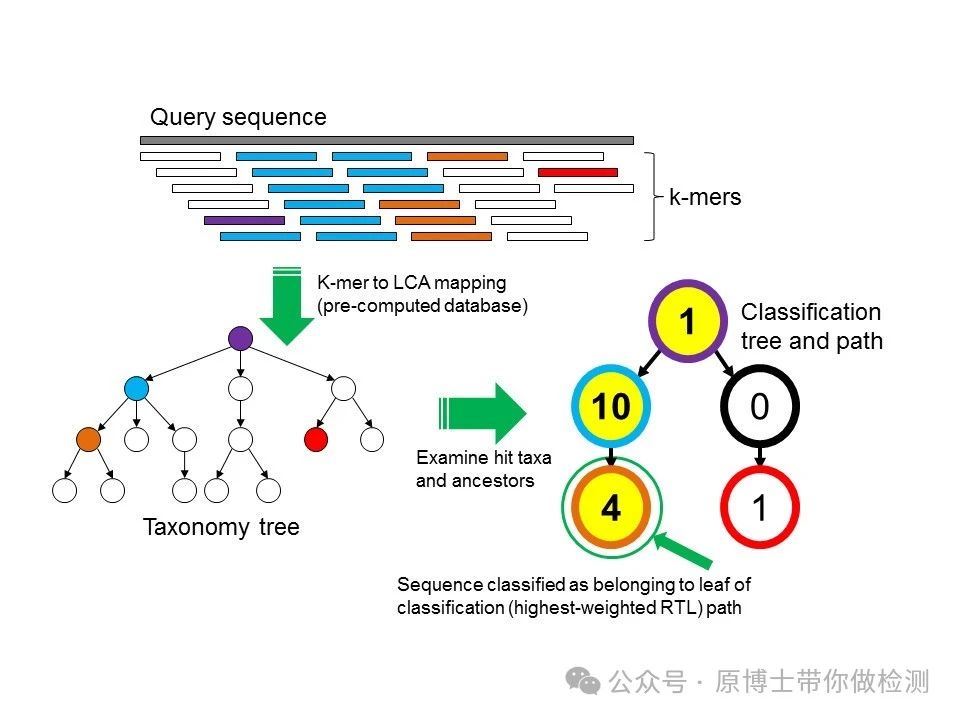
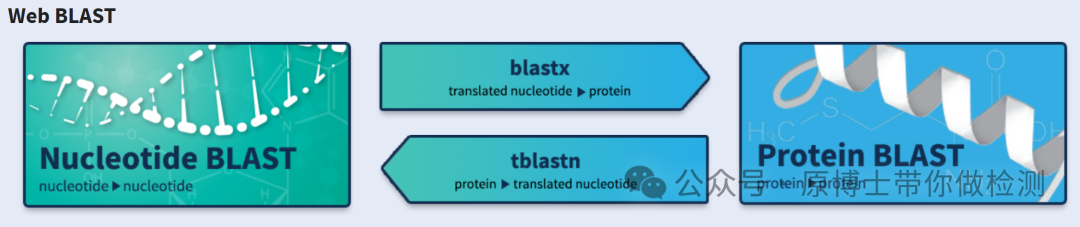
3.2.3 物种鉴定 动物病原微生物宏基因组高通量测序实验室应建立完善的动物病原微生物生物信息学分析流程,包括序列比对、物种鉴定等。微生物种类鉴定应至少鉴定到具体微生物种属。常用软件有:Kraken2、Centrifuge、BLAST等。 Kraken 2 : 是Kraken 的最新版本,Kraken 是一种分类系统 ,使用精确的 K-MER 匹配来实现高精度和快速分类速度。此分类器将查询序列中的每个 k-mer 匹配到最低值 包含给定 K-mer 的所有基因组的共同祖先 (LCA)。 Centrifuge:一种非常快速且节省内存的系统,用于对微生物样品中的 DNA 序列进行分类,其灵敏度高于其他领先系统,准确度也相当。该系统使用基于 Burrows-Wheeler 变换 (BWT) 和 Ferragina-Manzini (FM) 指数的新型索引方案,专门针对宏基因组分类问题进行了优化。Centrifuge 需要相对较小的索引,但分类速度非常快,使其能够在一小时内处理典型的 DNA 测序运行。 BLAST : 可查找序列之间具有局部相似性的区域。该程序将核苷酸或蛋白质序列与序列数据库进行比较,并计算匹配的统计显著性。BLAST 可用于推断序列之间的功能和进化关系,并帮助识别基因家族的成员。
Kraken分析流程
BLAST网页界面 3.2.4 致病微生物识别 宏基因组高通量测序结果中可能含有多种微生物信息,例如正常菌群、环境微生物和污染微生物。在动物病原微生物宏基因组高通量测序中,应结合临床症状对致病性微生物进行识别。
建议建立动物致病病原微生物数据库,用于动物病原微生物宏基因组高通量测序报告的判定指导,排除非病原微生物对结果判读的影响。
中科基因病原微生物信息分析系统 宏基因组高通量测序获得的微生物序列数(Reads)受测序样品量、样品质量、下机数据量等因素影响,每个样品测序获得的微生物序列数不一致。因此宏基因组高通量测序结果判定时,建议将测序获得的微生物序列数进行归一化,以每百万条序列数(Reads per million,RPM)或每千万条序列数(Reads per ten million,RPTM)为单位。 序列数(Reads):比对到特定物种属或种特异序列条数。 每百万序列数(reads per million,RPM):每百万测得序列中比对到目标物种基因组的序列条数。 每千万序列数(reads per ten million,RPTM):每千万条序列中比对到目标物种基因组的序列条数。
各实验室应以归一化后的微生物序列数、物种覆盖度或相对丰度为基础,结合阴性对照、测序样品信息(样品类型、采样部位、是否污染)和临床症状表现制定相应的结果判定标准。 覆盖度(Coverage): 检测到某种微生物的序列覆盖到该微生物基因组参考序列的比值,覆盖度越高表示检测到这种微生物基因组次数越多,可信度就越高。一般用%表示。 测序深度(Depth):将能与基因组比对上的序列碱基数累加并除以基因组被覆盖区域的总长,数值越大表示样本中检测到目标基因组的次数越多,可靠性也就越高。一般用1X、10X、100X表示。 相对丰度(Relative Abundance):指注释到该物种的序列数占样本中所有微生物总序列数的百分比(不含宿主)。丰度越高表示其在相同类型微生物中所占的比例越高,可信度也就越高。 绝对丰度(Absolute Abundance):指注释到该物种的序列数占总数据量的百分比(含宿主)。
建议阈值为检出3条微生物序列数,且能覆盖3个非重复区域。 宏基因组结果的判定比较复杂,目前行业内还没有形成统一的共识,有些文献将阈值设定为3-30 /1M reads,不建议一刀切的以几条序列数作为判定依据。判定的目的是确认测序结果中存在某物种。 对于二代测序结果,由于序列短,容易出现假阳性的比对结果,因此需要保证测序的序列数覆盖3个非重复区域。 对于三代测序结果,如果获得较长序列(500bp以上),1条序列理论上也可以确定,但是3条以上更加可靠。
17条序列都在同一位置的重复序列假阳性结果 5.1 数据质量控制 5.1.1 文库质量控制 实验室应根据测序目的、测序方法、测序长度等因素选择合适的建库试剂。每批文库都应进行质量评估,插入片段大小、单个文库浓度、混合文库浓度等指标符合测序仪上机要求后才能再进行下一步测序。 5.1.2 测序数据质量控制 动物病原微生物宏基因组高通量测序数据,建议 二代测序碱基识别质量Q30≥90%,每个样品的序列数≥10 M(10 million); 三代测序Q10≥80%,每个样品的数据量≥1Gb(1 Gbase)。 对于宏基因组测序,二代测序有比较明确的标准,一般医学临检要求序列数达到10-20M以上。三代测序的Q值和数据量目前尚没有明确的标准,还需要数据来验证,这里的标准为临时性标准,比二代测序标准低,此处建议为结合目前仪器性能的最低标准。 部分实验室为了降低测序成本和客户使用成本,随意进行合样检测(30合1甚至50合1),目的是降低测序成本,代价是降低测序数据量和检测敏感性,引起假阴性(漏检)检测结果。
5.1.3性能验证 宏基因组高通量测序需要通过质控品和临床样品对全流程的湿实验和干实验进行性能验证,确认方法的符合率、检出限、重复性、准确性等性能参数,符合实验室对病原微生物测序能力的要求。 5.2 阴性对照 宏基因组高通量测序实验各个环节中都可能产生气溶胶污染,每批实验都应加入阴性对照,与样品一起参与核酸提取、文库制备、上机测序和数据分析,监控整个实验过程。 建议阴性对照选用无支原体和外源病毒污染的洁净细胞,细胞密度至少为105 cells/mL。也可选择异源或远源物种的核酸(如拟南芥等模式植物、DNA噬菌体、RNA噬菌体等)做阴性对照,监控实验过程。
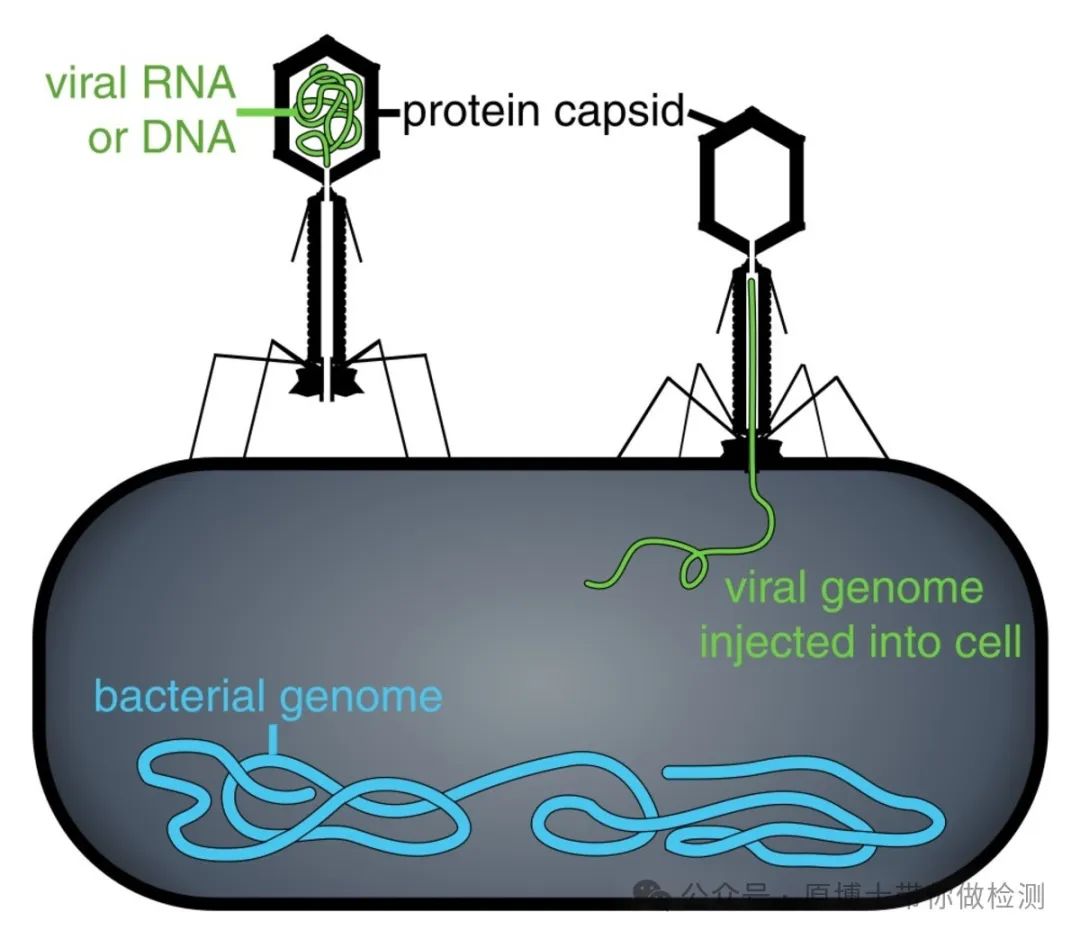
噬菌体示意图 5.3 阳性质控品 当测序平台或测序方法改变时,应使用阳性质控品对新的测序平台或方法进行性能验证。各实验室可选用已知阳性样品、病毒/菌培养物或者商品化动物病原微生物质控品作为阳性质控品使用。 阳性质控品应至少包含DNA病毒、RNA病毒、革兰氏阳性菌、革兰氏阴性菌、真菌、寄生虫共6类病原微生物类型。 如果一个实验室声明其所开展的宏基因组测序可以覆盖DNA病毒、RNA病毒、革兰氏阳性菌、革兰氏阴性菌、真菌、寄生虫等6类病原微生物类型。则应提供其验证数据,至少应包含对每种微生物的最低检测限和重复性结果。
中科基因高通量测序阳性质控盘 6.1 报告内容 宏基因组高通量测序报告中应包含物种信息、样品类型、临床症状及初步诊断、微生物中文名称、拉丁文名称、微生物序列数、测序覆盖度及相对丰度、质控数据等关键信息,对报告中出现的专业术语、技术参数进行注释。 临床病原微生物诊断时,为指导至病性病原微生物诊断及防治方案制定,建议对重要的病原微生物进行进一步注释,包括临床症状、易感动物、传播途径、疫病分类等。
6.2 报告解读 因病程差异、病原微生物个体差异、测序策略的局限等原因,动物病原微生物宏基因组高通量测序结果须结合患病动物临床症状和流行病学信息对测序结果进行报告解读。 宏基因组高通量测序技术能够识别多种病原微生物, 在动物病原微生物检测中具有显著优势。然而,在实际应用中,由于宏基因组高通量测序技术流程复杂,包括样品采集与保存、核酸提取、文库制备、实验污染控制、生物信息学分析、结果解读与报告、质量控制与标准化等各方面对技术平台和人员的专业知识和技能要求高,运行难度大。本共识希望通过不断完善、优化宏基因组高通量测序技术流程和应用策略,指导动物病原微生物检测行业更好发展。
宏基因组测序技术的应用方向 文献13 原博士小结: 这篇专家共识的编写汇集了国内兽医领域开展mNGS测序的上游仪器厂家、高校科研院所、动物疫控中心、检测机构和研发企业的资深专家。在编写过程中有很多内容存在着一定的争议,有的专家觉得过于严格,有的专家认为标准太低,但是由于目前国内缺乏mNGS标准,一个存在争议的专家共识对行业也是利大于弊的。我们期望大家能够对这个专家共识进行广泛讨论甚至批评,让真理越辩越明,也让这个2024版的专家共识更快的升级迭代。 参与本共识编写的人员及单位: 参考文献: [1]宏基因组分析和诊断技术在急危重症感染应用专家共识组,童朝阳,宋振举.宏基因组分析和诊断技术在急危重症感染应用的专家共识[J].中华急诊医学杂志,2019,28(2):151-155 [2]张述耀,侯铁英,黎小妍,等.纳米孔测序在病原微生物检测中的应用专家共识[J].中国药房,2024,35(14):1673-1682. [3]世界动物卫生组织(WOAH).WOAH陆生动物诊断试验与疫苗手册(哺乳动物、禽类与蜜蜂)第1卷 [M].北京:中国农业出版社,2022:12-20,84-89 [4]国家卫生健康委. 国家卫生健康委关于印发人间传染的病原微生物目录的通知 [EB/OL].(20230818)[2024-07-31].http://www.nhc.gov.cn/cms-search/xxgk/getManuscriptXxgk.htm?id=b6b51d792d394fbea175e4c8094dc87e [5]中国药师协会,中华医学会细菌感染与耐药防治分会,国家卫生健康委临床抗微生物药物敏感性折点研究和标准制定专家委员会.病原宏基因组高通量测序临床本地化检测规范专家共识[J].中华预防医学杂志, 2024, 58(04):454-465.DOI:10.3760/cma.j.cn112150-20230720-00019. [6] 农业农村部.中华人民共和国农业农村部公告 第573号 [EB/OL].(2022-06-29)[2024-07-31]. https://www.moa.gov.cn/govpublic/xmsyj/202206/t20220629_6403635.htm [7] GB 19489-2008.实验室 生物安全通用要求[S] [8] GB/T 27401-2008.实验室质量控制规范 动物检疫[S] [9]国务院.病原微生物实验室生物安全管理条例 [EB/OL].(2018-03-19)[2024-07-31]. http://www.gov.cn/gongbao/content/2019/content_5468882.htm [10]病原宏基因组高通量测序临床本地化检测规范专家共识[J]. 中华预防医学杂志,2024,58(04):454-465.DOI:10.3760/cma.j.cn112150-20230720-00019 [11]中华医学会检验医学分会.高通量宏基因组测序技术检测病原微生物的临床应用规范化专家共识[J].中华检验医学杂志, 2020,43(12):1181-1195.DOI:10.3760/cma.j.cn114452-20200903-00704. [12]中华医学会检验医学分会.宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识[J].中华检验医学杂志, 2021, 44(9):9.DOI:10.3760/cma.j.cn114452-20210322-00178. [13]Chiu, C.Y., Miller, S.A. Clinical metagenomics. Nat Rev Genet 20, 341–355 (2019). [14]Tianyuan Zhang, Hanzhou Li, Mian Jiang, Huiyu Hou, Yunyun Gao, Yali Li, Fuhao Wang, Jun Wang, Kai Peng, Yong-Xin Liu. (2024). Nanopore sequencing: flourishing in its teenage years. Journal of Genetics and Genomics. |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号