5月10日,上海市卫健委发布了《上海市新生儿遗传代谢病筛查技术规范(2024版)》。
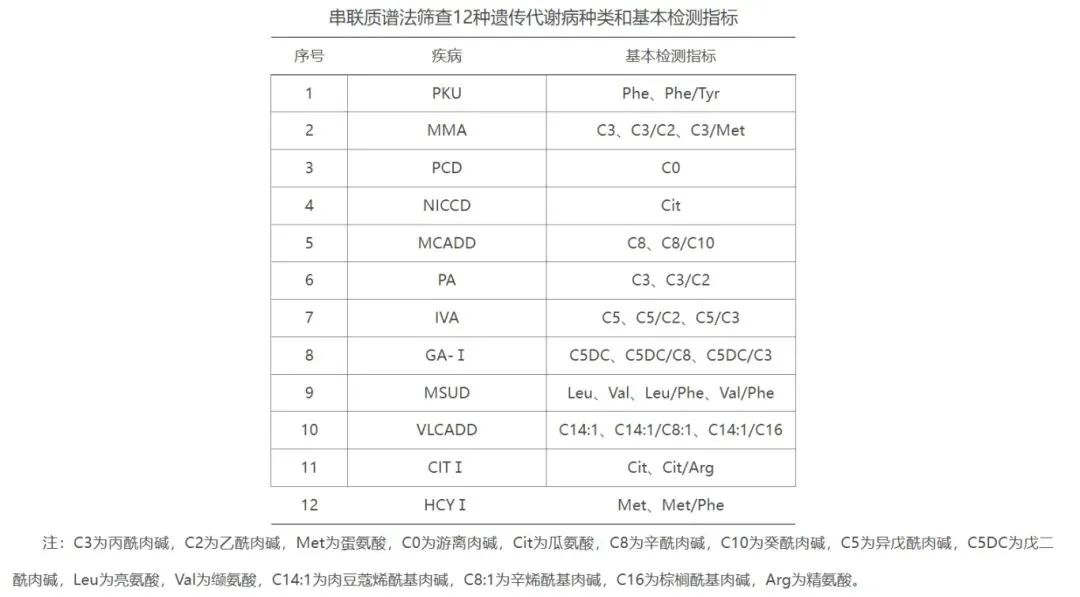
原文 上海市新生儿遗传代谢病筛查技术规范 (2024版) 一、遗传代谢病的定义 遗传代谢病是由于基因改变导至代谢途径中相关酶或蛋白功能下降或缺乏,引起某些特异代谢物增高或降低,造成机体损伤的一大类疾病,可通过检测这些特异代谢物,筛查或协助诊断相关疾病。 二、新生儿遗传代谢病筛查 新生儿遗传代谢病筛查是指在新生儿群体中,通过采集其血液对一些严重危害儿童身心健康的先天性、遗传代谢病进行检测,以期在临床症状出现前或发病早期及时给予治疗,避免或减轻患儿机体各系统受到不可逆的损害。 本市确定对先天性甲状腺功能减低症(CH)、苯丙酮尿症(PKU)、先天性肾上腺皮质增生症(CAH)、葡萄糖-6-磷酸脱氢酶缺乏症(G6PD)、甲基丙二酸血症(MMA)、原发性肉碱缺乏症(PCD)、希特林蛋白缺乏症(NICCD)、中链酰基辅酶A脱氢酶缺乏症(MCADD)、丙酸血症(PA)、异戊酸血症(IVA)、戊二酸血症-Ⅰ型(GA-Ⅰ)、枫糖尿病(MSUD)、极长链酰基辅酶A脱氢酶缺乏症(VLCADD)、瓜氨酸血症Ⅰ型(CITⅠ)和同型半胱氨酸血症Ⅰ型(HCYⅠ)等15种疾病开展全市性筛查。 三、血片采集 (一)机构设置 本市开展的新生儿遗传代谢病筛查采血工作由本市各级各类经批准开展助产技术服务以及接受新生儿转诊的医疗机构承担。 (二)人员要求 必须具有与医学相关的中专以上学历,并从事医学临床工作2年以上,同时接受过相关知识和技能的培训并取得合格证书。 培训内容包括新生儿遗传代谢病筛查的目的、原则、方法以及网络运转,血片采集、保存、递送相关知识,相关信息和档案管理等内容。 (三)血片采集步骤 1.血片采集人员清洗双手并佩戴无菌、无滑石粉的手套。 2.按摩或热敷新生儿足跟,选择足跟内侧或外侧缘并用75%乙醇消毒皮肤。 3.待乙醇完全挥发后,使用一次性采血针针刺消毒部位,深度小于2毫米,挤压局部使流出的血液呈“水滴”状,用干棉球拭去第1滴血,从第2滴血开始留取标本。 4.将滤纸片接触血滴,切勿触及足跟皮肤,使血液自然渗透至滤纸背面,避免重复滴血,根据筛查病种,至少采集5个血斑。 5.手持消毒干棉球轻压采血部位止血。 6.若新生儿需采集静脉血进行其他检测项目,可把注射器内静脉血滴于滤纸上制作血片,不需另外采集足跟血。 7.将血片悬空平置,自然晾干呈深褐色。避免阳光及紫外线照射、烘烤、挥发性化学物质等污染。 8.及时将检查合格的滤纸干血片置于有干燥剂的密封袋内,密闭保存在2~8℃冰箱中,有条件者可在0℃以下保存。 9.所有采集的血片应当按照血源性传染性标本对待,对特殊传染病(如艾滋病等)患儿标本,应予标识并单独包装。 (四)质量标准 1.血片采集的滤纸应当选择新生儿筛查专用滤纸。 2.采血针必须一人一针,使用后的采血针应按利器医疗废物处理。 3.正常采血时间为出生后48h~7d,婴儿应充分哺乳。 4.因各种原因(早产、低出生体重、正在接受治疗、提前出院等)未能及时采血者,最迟采血时间不宜超过出生后20天;对于输血治疗的新生儿,应在输血治疗前完成第一次血片采集,输血治疗120天后再次进行血片采集;对于拟行骨髓移植或干细胞移植的新生儿应在治疗前完成采样。 5.合格滤纸干血片应为:(1)滤纸血斑个数满足检测需求,每个血斑直径大于8毫米;(2)血滴自然渗透,滤纸正反面血斑一致;(3)血斑无污染;(4)血斑无渗血环;(5)有完整的血片采集信息记录。 6.血样标本应当在采集后24小时内递出。 7.采血机构应当保存的资料包括:血样标本登记单、与递送机构的血样标本交接签收单、反馈的检测及确诊结果等,保存期限至少10年。 血样标本登记内容包括:采血单位、血片编号、母亲住院号、母亲姓名、户籍、孕周、婴儿姓名、婴儿性别、出生体重、出生日期、家庭地址、邮编、联系电话、采血日期、采血者等。 四、实验室检测 (一)机构要求 取得《医疗机构执业许可证》,经市卫生健康委遴选认定。 (二)人员要求 业务负责人:与医学相关的本科及以上学历,高级职称,具有儿科或临床检验工作经验,从事新生儿疾病筛查工作5年以上,掌握新生儿遗传代谢病筛查网络运作和管理技能。 实验室技术人员:中专或以上学历,从事检验工作2年以上,具有技师或以上技术职称,接受过新生儿遗传代谢病筛查相关知识和技能培训。 文员:熟练掌握计算机操作(文字处理及统计、信息系统管理)技术且有档案管理工作经验。 (三)设备及试剂要求 根据筛查病种及检测数量配备相应的设备,主要包括酶标仪、荧光分析仪或全自动荧光分析仪及液相串联质谱分析系统等。 采用国家规定实验方法,且具有国家批准文号的试剂和设备进行检测。 (四)房屋要求 1.实验室用房2间以上,至少60平方米; 2.综合用房2间,至少20平方米以上,用于滤纸干血片样本的验收、临床信息录入和资料登记保存; 3.储藏室或冷库(2~8℃)1间,用于滤纸干血片的保存;如长期(>5年)储存,建议在-20℃条件下保存滤纸干血片。 (五)检测技术方法 采用荧光法或免疫荧光法筛查CH、CAH、G6PD等3种疾病。 采用串联质谱法筛查PKU、MMA、PCD、NICCD、MCADD、PA、IVA、GA-Ⅰ、MSUD、VLCADD、CITⅠ、HCYⅠ等12种疾病。 1.PKU (1)以滤纸干血片苯丙氨酸(Phe)含量作为筛查指标。 (2)Phe>120μmol/L及Phe与酪氨酸(Tyr)比值(Phe/Tyr)>2.0作为筛查阳性。 (3)早产儿、未成熟儿、肝损害患儿可出现高苯丙氨酸血症(HPA)假阳性,可通过肝功能检查、血尿代谢物质谱分析和基因分析进行鉴别;标本采集时若受检者蛋白质负荷不足,可出现假阴性。 2.CH (1)以滤纸干血片促甲状腺激素(TSH)作为筛查指标。 (2)TSH阳性切值根据实验室及使用的试剂盒确定,一般为8~10μIU/ml,高于切值为筛查阳性。 (3)低出生体重儿或极低出生体重儿可在生后2~4周或体重超过2500g时重新采血复查,以减少首次筛查可能出现的假阴性。 (4)以TSH为筛查指标,可筛查因甲状腺缺如、萎缩或发育不良导至的原发性CH,对垂体或下丘脑功能低下引起的继发性CH或TSH延迟升高者可能出现假阴性,筛查漏诊率5%~10%,故对有疑问的样本,需重新采血送检,可直接进行血清甲状腺功能检测。 3.CAH (1)以滤纸干血片17-羟孕酮(17-OHP)作为筛查指标,目标疾病主要为经典型21-羟化酶缺乏导至的CAH。 (2)17-OHP阳性切值根据实验室及使用的试剂盒确定,正常分娩新生儿一般为10~40nmol/L,高于切值为筛查阳性。因17-OHP含量受胎龄影响较大,可建立不同胎龄新生儿的阳性切值。 (3)应激反应、早产、低出生体重、危重疾病、黄疸、脱水等易导至CAH筛查假阳性;孕母或新生儿糖皮质激素治疗史易导至CAH筛查假阴性。 4.G6PD (1)以滤纸干血片G6PD活性作为筛查指标。 (2)G6PD活性切值一般为2.0~2.6U/gHb或15~30U/dl,低于切值为筛查阳性。 (3)以G6PD活性为筛查指标对女性杂合子不敏感,易出现假阴性。G6PD活性检测结果易受血片采集、递送时间、温度和湿度、标本保存条件等影响而出现假阳性,夏季运输过程中血片需保持低温。 5.串联质谱法筛查的多种遗传代谢病 (1)筛查指标为氨基酸、游离肉碱及酰基肉碱。 (2)样本前处理可采用衍生化法和非衍生化法。 (3)不同疾病的筛查指标浓度及比值的阳性切值根据各实验室及使用的试剂盒确定。 (4)早产、低体重、高蛋白饮食、抗生素使用、产妇疾病以及疾病指标的敏感性等均会影响筛查结果,出现假阳性或假阴性。
(六)质量控制 1.收到标本应当在24小时内登记,不符合要求的标本应当立即退回重新采集。5个工作日内出具报告,对阳性标本要进行原标本复查,如仍阳性者,再出具阳性报告。 2.每月向开展新生儿遗传代谢病筛查血片采集的机构或助产机构反馈实验室检测结果,阳性检测结果应及时反馈。 3.实验室应制定切实可行的质控程序,选择合适的质控规则,并在日常实验中参照执行,以监控潜在的系统误差和随机误差。 4.实验室应定期进行仪器校准,试剂更换批次时进行比对验证,以保证检测平台稳定、受控,检测结果可靠。 5.每月召开实验室质控会议,针对实验室质量指标进行分析讨论。质量指标包括:(1)检验前的新生儿遗传代谢病健康教育知晓率、检测血片不合格率;(2)检验中的室内质控开展率、变异系数不合格率、室间质评参加率;(3)检验后的初筛阳性召回率、阳性预测值、检测病种发病率及接到标本5个工作日内出报告率。 6.每年参加国家卫生健康委员会临床检验中心新生儿遗传代谢病筛查实验室室间质量评价,并且成绩合格。 7.血片标本必须在8℃以下保存至少5年,以备复查。 8.有完整的实验室检测资料和信息安全保障措施,资料存档保留至少10年。资料包括:(1)每份血样标本的新生儿的信息;(2)不符合要求退回标本,并注明原因及日期;(3)每次实验结果的原始资料,包括标准曲线、质控结果、筛查结果;(4)有关质量控制资料,包括室内质控图、室间质评反馈、失控原因、纠正方法等;(5)筛查阳性需及时通知追踪随访机构,并作相应记录。 (七)规章制度 1.人员分工责任制度; 2.各种技术操作常规; 3.质量控制管理制度; 4.仪器管理及校准制度; 5.试剂材料管理制度; 6.血样标本登记保存制度; 7.安全制度。 五、诊断及干预 (一)基本要求 1.机构设置:承担新生儿遗传代谢病诊治工作的新生儿遗传代谢病筛查中心或具有能力的医疗机构,应由市卫生健康委根据本行政区域的实际情况遴选认定。 2.人员要求:(1)承担新生儿遗传代谢病筛查阳性召回工作的人员,应当具有与医学相关的中专以上学历,从事医疗保健工作2年以上;(2)从事诊治的人员必须取得执业医师资格,具有中级以上儿科临床专业技术职称,通过遗传代谢病、内分泌等专业与新生儿遗传代谢病筛查及诊治相关知识和技能培训。 3.诊治机构与人员职责:严格遵循新生儿疾病筛查技术规范的要求,建立专科档案与管理制度、召回制度、转诊制度、随访评估制度。对筛查阳性者应召回,提供进一步的确诊或鉴别诊断服务,并对确诊患儿进行相应治疗。做好统计、分析、上报和反馈筛查阳性者的确诊、治疗及评估情况。 (二)筛查阳性召回 1.依托我市目前较为完善的妇幼保健三级网络对新生儿遗传代谢病筛查阳性者进行召回。 2.负责召回的人员接到阳性报告后,应采用各种方式立即通知监护人带筛查阳性婴儿到新生儿遗传代谢病筛查中心及时复查。 3.因地址不详或拒绝随访等原因失访者,须注明原因,做好备案工作。每次通知均须详细记录。 (三)诊断及治疗 诊断原则:(1)新生儿筛查阳性者,根据不同疾病选择相关的检测。(2)根据筛查指标异常程度,可只采血片复查,或进入确诊程序,进行相关实验室检测。(3)筛查阳性者经上述检测提示相应遗传代谢病,建议进行鉴别诊断和基因诊断,明确相关基因的致病性变异位点。(4)特异性生化指标显著异常者,即使没有基因检测结果,或基因检测未明确致病性变异位点,仍可诊断。 治疗原则:(1)筛查阳性新生儿一旦确诊,需要尽快治疗,治疗越早,疗效越好。(2)指标显著异常新生儿,在进行相关实验室检测的同时,应立即进行治疗。(3)治疗原则为降低体内与疾病相关代谢途径的前体物质及其旁路代谢产物、补充缺乏的产物,减轻这些病理改变对机体造成的损害。(4)治疗方法依据疾病不同和疾病严重程度而不同,包括饮食治疗、药物治疗、透析治疗、器官及细胞移植治疗、康复治疗等。(5)需要饮食治疗的代谢病,治疗过程中根据疾病特点定期监测血氨基酸浓度、肉碱浓度,避免这些物质含量过低或过高对机体造成危害。(6)筛查确诊的患者大部分需要终生治疗,治疗过程中需要定期随访,监测体格、智力发育水平和代谢状况。 随访原则:(1)筛查确诊患儿需要坚持长期随访、规范随访。(2)随访频次根据疾病和病情需要确定,建议0~3月龄每个月随访一次,4~12月龄每3个月随访一次,1岁以后每3~6个月随访一次。 本市新生儿遗传代谢病筛查的疾病包含15种。 1.PKU (1)筛查阳性召回标准 Phe浓度>120μmol/L和Phe/Tyr>2.0。 (2)诊断 血Phe浓度持续>120μmol/L和血Phe/Tyr>2.0称为HPA。所有HPA患儿建议进行尿蝶呤谱分析、血二氢蝶啶还原酶(DHPR)活性测定和基因检测,以鉴别苯丙氨酸羟化酶(PAH)缺乏症和四氢生物蝶呤(BH4)缺乏症。BH4负荷试验可协助诊断。 ①PAH缺乏症分型:血Phe≥1200μmol/L为经典型PKU;血Phe 360~<1200μmol/L为轻度PKU;血Phe>120~<360μmol/L为轻度HPA。 ②BH4缺乏症分型:最常见6-丙酮酰四氢蝶呤合成酶缺乏症(尿新蝶呤增高,生物蝶呤及生物蝶呤与新喋呤比例降低),其次为DHPR缺乏症(DHPR活性明显降低),其他类型少见。 (3)治疗 ①PAH缺乏症:初筛血Phe>360μmol/L,或初筛Phe>120~<360μmol/L者,间隔一周血Phe浓度>360μmol/L均应给予低Phe饮食治疗,血Phe浓度≤360μmol/L者暂不需要治疗。治疗与不需要治疗的患儿均需定期监测血Phe浓度:低Phe饮食治疗者,如血Phe浓度异常,每周检测一次;如血Phe浓度控制在理想范围内,每月检测1~2次。儿童血Phe理想范围:0~1岁120~240μmol/L,1~12岁120~360μmol/L,>12岁120~600μmol/L。定期进行体格发育评估,在1岁、2岁、3岁、6岁时进行智能发育评估。提倡终生治疗。成年PKU女性患者应在怀孕前半年起严格控制血Phe浓度在120~360μmol/L,直至分娩。 ②BH4缺乏症:按不同病因给予BH4或低Phe饮食治疗、补充神经递质前体(美多巴、5-羟色氨酸)等联合治疗。 2.CH (1)筛查阳性召回标准 血TSH>8~10μIU/ml。 (2)诊断 确诊指标为血清TSH和游离甲状腺素(FT4)浓度。TSH增高、FT4降低为CH;TSH增高、FT4正常为高TSH血症;TSH正常或降低、FT4降低为中枢性甲状腺功能减低症。 (3)治疗 ①甲状腺激素替代治疗:确诊患儿口服左旋甲状腺素(L-T4),初始剂量为5~15μg/(kg·d),每日一次,使FT4在2周内、TSH在4周内达到正常。 ②高TSH血症:TSH>10mIU/L,给予L-T4治疗。TSH 6~10mIU/L,建议间隔1个月复查甲状腺功能,如长期维持在6~10mIU/L之间,可给予小剂量L-T4替代治疗。 ③定期复查甲状腺功能:初次治疗后2周复查,根据血FT4、TSH浓度调整治疗剂量,使血FT4维持在平均值至正常上限范围内,TSH维持在正常范围内。甲状腺功能1岁内每2~3个月复查一次,1岁以上3~4个月复查一次,3岁以上每6个月复查一次。 ④定期体格发育评估,在1岁、2岁、3岁、6岁时进行智能发育评估。 ⑤CH患儿随访过程中依据甲状腺功能检测结果调整L-T4剂量,部分患儿需要终生治疗,其他患儿可逐渐减量,直至停药。停药患者随访1年,甲状腺功能仍正常者可终止随访。 3.CAH (1)筛查阳性召回标准 血17-OHP大于筛查实验室的切值。由于血17-OHP筛查切值与新生儿胎龄有关,各筛查实验室需要建立不同胎龄的切值。 (2)诊断 ①血17-OHP浓度持续增高;②血雄烯二酮、睾酮增高;③血促肾上腺皮质激素增高或正常;④失盐型血钠降低,血钾增高;⑤基因检测到变异位点;⑥临床表现包括皮肤色素沉着,女性患儿**增大。符合①、②、③或①和⑤可以诊断。 (3)治疗 一旦确诊为经典型21-羟化酶缺乏症,立即开始肾上腺皮质激素替代治疗。 ①氢化可的松:开始剂量可偏大,25~50mg/(㎡·d),以尽快控制代谢紊乱。临床症状好转、电解质正常后则尽快将药物减少至维持量,婴儿期8~12mg/(㎡·d),分3次,每8h口服一次,以后根据临床及检测指标调整剂量。 ②盐皮质激素:经典型(失盐型及单纯男性化型)患儿,尤其在新生儿期及婴儿早期,均需同时给予盐皮质激素,以改善失盐状态。9α-氟氢可的松0.1~0.2mg/d,分2次口服,通常治疗数日后电解质水平趋于正常,维持量为0.05~0.1mg/d。失盐型患儿每日补充氯化钠l~2g。 ③应激状态处理:在发热超过38.5℃、腹泻伴脱水、全麻手术和危象发生,或危重情况下,可增加氢化可的松剂量至50~100mg/(㎡·d)。 ④外生殖器矫形治疗:对**肥大及**融合的女性患儿,在代谢紊乱控制后,应在出生后3~12个月尽早实施整形手术。 ⑤随访:治疗初期需密切随访,每2周至1个月随访一次。代谢稳定后,≤2岁每3个月一次;>2岁每3~6个月一次。每3~6个月测量身长/身高,每年评估骨龄。氢化可的松剂量调节的重要指标为17-OHP、雄烯二酮、睾酮及生长速度。 4.G6PD (1)筛查阳性召回标准 血G6PD活性低于筛查实验室切值。 (2)诊断 ①血G6PD活性或G6PD/6PGD比值降低。 ②G6PD基因检测到变异位点。 (3)治疗及预防 ①治疗:无症状时无需治疗。若出现溶血症状,根据胆红素及贫血程度,给予降胆红素治疗,贫血严重者输入G6PD活性正常的血液治疗。 ②预防:避免食用蚕豆及其制品,避免接触樟脑丸等日常用品,避免使用氧化型药物,包括磺胺甲氧嗪、磺胺吡啶、磺胺异恶唑、呋喃唑酮、安痛定、阿司匹林等药物。 5.NICCD (1)筛查阳性召回标准 血瓜氨酸(Cit)增高,伴或不伴有甲硫氨酸(Met)、Phe、Tyr及精氨酸(Arg)增高。 (2)诊断 ①血Cit增高,伴或不伴有Met、Arg、Tyr及Phe增高,部分患儿伴有多种酰基肉碱增高。 ②尿4-羟基苯乳酸及4-羟基苯丙酮酸增高。 ③其他生化检测:甲胎蛋白显著增高,直接胆红素、总胆汁酸和肝酶升高;血糖降低、白蛋白降低及凝血功能障碍。 ④SLC25A13基因检测到变异位点。 (3)治疗 给予无乳糖富含中链甘油三脂配方营养粉,补充脂溶性维生素,多数症状可在1岁内缓解。 6.MSUD (1)筛查阳性召回标准 血亮氨酸(Leu)、Leu/Phe增高,伴或不伴缬氨酸(Val)、Val/Phe增高。 (2)诊断 ①血Leu、Val、Leu/Phe及Val/Phe增高。 ②尿2-羟基异戊酸、2-酮异戊酸、2-酮-3-甲基戊酸、2-酮异己酸增高。 ③BCKDHA、BCKDHB、DBT或DLD基因检测到变异位点。 (3)治疗 ①急性期治疗:限制蛋白质摄入,给予不含Leu、异亮氨酸及Val的特殊营养粉治疗,静脉滴注左卡尼汀、葡萄糖及脂肪乳剂供能。试用维生素B1 100~300mg/d,有效者Leu可下降50%以上。 ②稳定期治疗:限制蛋白质摄入,给予不含Leu、异亮氨酸及Val的特殊营养粉,保证足够热量和营养,定期检测支链氨基酸浓度及体格和智力发育;药物治疗:左卡尼汀及维生素B1(有效患儿)。 ③肝移植:病情严重、反复发作者可考虑肝移植,但不能逆转慢性脑损伤。 7.HCYⅠ (1)筛查阳性召回标准 血Met及Met/Phe增高。 (2)诊断 ①血同型半胱氨酸(HCY)增高。 ②血Met及Met/Phe增高。 ③CBS基因检测到变异位点。 (3)治疗 ①限制天然蛋白质摄入量,Met增高患儿给予无Met特殊营养粉饮食。 ②药物治疗:甜菜碱 100~250mg/(kg·d)、维生素B6 300~600mg/d(少数患儿有效)、叶酸5~10mg/d及甲钴胺1mg/d。 8.CITⅠ (1)筛查阳性召回标准 血Cit、Cit/Arg增高。 (2)诊断 ①血Cit、Cit/Arg增高。 ②尿乳清酸和尿嘧啶增高或正常。 ③ASS1基因检测到变异位点。 (3)治疗 ①急性期治疗:限制蛋白质摄入;降血氨,应用苯甲酸钠、Arg及左卡尼汀,严重者需要血液透析或者腹膜透析治疗;纠正水电解质紊乱,静脉滴注葡萄糖。 ②稳定期治疗:低蛋白饮食,保证热量、维生素和微量元素需要量;长期应用Arg、苯丁酸钠及苯甲酸钠等药物治疗。 9.MMA (1)筛查阳性召回标准 ①血丙酰肉碱(C3)、C3/乙酰肉碱(C2)均增高。 ②血C3正常,C3/C2或C3/Met增高。 ③仅血C3增高,但C3/C2接近正常切值高限。 (2)诊断 ①血C3及C3/C2增高,或者仅有C3/C2增高。合并型MMA常伴有Met水平下降,C3/Met增高。 ②尿甲基丙二酸增高,可伴有甲基枸橼酸、3-羟基丙酸及丙酰甘氨酸增高。 ③合并型MMA患儿血HCY增高。 ④基因检测:单纯型MMA检测MMUT、MMAA、MMAB、MCEE、SUCLG1及SUCLA2等基因;合并型MMA检测MMACHC、MMADHC、LMBRD1、HCFC1及ABCD4等基因。 (3)治疗 ①急性期治疗:限制天然蛋白质摄入或给予不含异亮氨酸、Val、Met及苏氨酸的特殊营养粉,补充葡萄糖及脂肪乳供能,纠正酸中毒及电解质紊乱,静脉滴注左卡尼汀。依据年龄给予维生素B121~5mg肌肉注射,每日一次,连续5d,用药前后进行血C3、C3/C2、尿有机酸检测,判断维生素B12是否有效。 ②稳定期治疗:a)单纯型MMA可分为维生素B12无效型和维生素B12有效型。维生素B12无效型单纯型MMA患儿需要控制天然蛋白质摄入量,给予不含异亮氨酸、Val、Met及苏氨酸的特殊营养粉饮食。因长期单独使用特殊营养粉喂养的患儿可导至必需氨基酸缺乏,一般不超过5d,之后与天然蛋白质混合喂养。此外,还需要给予左卡尼汀50~200mg/(kg·d)治疗。维生素B12有效型单纯型MMA患儿需要饮食治疗及左卡尼汀治疗,同时给予维生素B12治疗。b)合并型MMA患儿均对维生素B12有效,且效果较好,可以不给予特殊饮食治疗,可给予维生素B12(羟钴胺效果优于其他类型的维生素B12)、左卡尼汀、甜菜碱、亚叶酸钙或叶酸等药物治疗,Met降低者给予Met治疗,并给予营养支持。 10.PA (1)筛查阳性召回标准 ①血C3和C3/C2均增高。 ②血C3正常,C3/C2增高。 ③仅血C3增高,但C3/C2接近正常切值高限。 (2)诊断 ①血C3及C3/C2增高,或者仅有C3/C2增高,可伴甘氨酸增高。 ②尿3-羟基丙酸及甲基枸橼酸增高,可伴有丙酰甘氨酸或甲基巴豆酰甘氨酸增高。 ③PCCA或PCCB基因检测到变异位点。 (3)治疗 ①急性期治疗:限制蛋白质摄入或补充去除异亮氨酸、Val、Met、苏氨酸的特殊营养粉;纠正酸中毒,静脉滴注左卡尼汀,补充葡萄糖、脂肪乳剂,补充液量及热卡。 ②稳定期治疗:限制天然蛋白质,补充去除异亮氨酸、Val、Met、苏氨酸的特殊营养粉或蛋白粉;避免治疗过度导至必需氨基酸缺乏;给予高热量饮食;左卡尼汀维持剂量为50~100mg/(kg·d)。 11.IVA (1)筛查阳性召回标准 血异戊酰肉碱(C5)及C5/C2、C5/C3增高。 (2)诊断 ①血C5及C5/C2、C5/C3增高。 ②尿异戊酰甘氨酸增高,可伴3-羟基异戊酸增高。 ③IVD基因检测到变异位点。 (3)治疗 ①急性期治疗:限制蛋白质饮食,给予不含亮氨酸的特殊营养粉,静脉滴注左卡尼汀,纠正水电解质紊乱,静脉滴注葡萄糖以提供热量。 ②稳定期治疗:限制天然蛋白质饮食,给予不含亮氨酸的特殊营养粉。补充甘氨酸100~600mg/(kg·d)和左卡尼汀50~200mg/(kg·d)。 12.GA-Ⅰ (1)筛查阳性召回标准 血戊二酰肉碱(C5DC)及C5DC/辛酰肉碱(C8)、C5DC/C3增高。 (2)诊断 ①血C5DC及C5DC/C8、C5DC/C3增高。 ②尿戊二酸显著增高。 ③GCDH基因检测到变异位点。 (3)治疗 ①饮食治疗:限制天然蛋白质,补充不含赖氨酸、色氨酸的特殊营养粉。 ②药物治疗:左卡尼汀50~200mg/(kg·d);维生素B2 50~200mg/d,少数患儿有效。 13.PCD (1)筛查阳性召回标准 ①血游离肉碱(C0)下降。 ②血C0正常,但接近正常值下限,其他酰基肉碱降低。 ③由于母源性肉碱缺乏较常见,建议召回新生儿同时采集母亲血进行串联质谱检测,以除外母源性肉碱缺乏。 (2)诊断 ①血C0下降,伴其他酰基肉碱降低,排除母源性肉碱缺乏。 ②SLC22A5基因检测到变异位点。 (3)治疗 ①避免过度饥饿和疲劳,防止低血糖发生。 ②补充左卡尼汀50~200mg/(kg·d)。 ③母源性肉碱缺乏:婴儿及母乳喂养的母亲均应口服左卡尼汀,婴儿血C0恢复正常后即可停药,母亲继续服用左卡尼汀。 ④多食富含左卡尼汀的食品,如牛肉及羊肉。 14.MCADD (1)筛查阳性召回标准 血C8、C8/葵酰基肉碱(C10)增高,伴或不伴己酰基肉碱及葵烯酰基肉碱增高。 (2)诊断 ①血C8升高显著,C8/C10比值增高。 ②ACADM基因检测到变异位点。 (3)治疗 治疗原则为避免低血糖、饥饿及劳累,急性发作期对症处理。继发性肉碱缺乏可适当补充左卡尼汀。 15.VLCADD (1)筛查阳性召回标准 血肉豆蔻烯酰基肉碱(C14:1)、C14:1/辛烯酰基肉碱(C8:1)及C14:1/棕榈酰基肉碱(C16)增高,伴或不伴其他酰基肉碱增高。 (2)诊断 ①血C14:1、C14:1/C8:1、C14:1/C16增高,伴或不伴其他酰基肉碱增高。 ②尿己二酸、辛二酸、癸二酸、十二烷二酸等水平升高,轻症可无二羧酸尿症。 ③ACADVL基因检测到变异位点。 (3)治疗 治疗原则是避免饥饿、长时间剧烈运动及劳累;高碳水化合物和低脂饮食,尤其是限制长链脂肪酸的摄入,补充中链甘油三酯。无症状无需饮食干预。肉碱降低者可适当补充左卡尼汀。 -END- |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号