IVD企业的出海合规路径:IVD或LDT模式?
2024-5-20 17:46|
发布者: 沙糖桔|
查看: 3772|
评论: 0|来源: 伙伴智能
摘要: 最终实现IVD产品化,可能是比较现实的路径。
| 2024 年 4 月29 日,美国食品药品监督管理局FDA 宣布了关于LDT的最终规章,并明确将在未来4年内逐步取消对LDT的一般执法自由裁量权。近期,也有些IVD企业咨询关于出海合规方面的路径选择:IVD认证抑或是LDT实验室模式?基于前期的了解,我们进行了相关总结,供相关企业参考。1、体外诊断(IVD,英文全称In-Vitro Diagnostics)指在人体之外,通过对人体样本(血液、体液、组织等)进行检测获取临床诊断信息,进而判断疾病或机体功能的产品和服务。2、实验室开发试验(LDT,英文全称Laboratory Developed Test)指针对尚未获得产品注册、仅在实验室内部研发、验证与使用的体外诊断项目,最早由美国提出并实践,主要是为了缓和医疗器械监管与实际临床需求之间的矛盾,允许满足特定条件的医学实验室自研并使用未经注册的体外诊断试剂产品用于临床诊断。FDA将 (LDT)定义为用于临床并在单个实验室内设计、制造和使用的IVD。LDT仅限所在医学检验部门内部使用,不得作为检测试剂出售给任何其它医学检验部门、医院及个人。 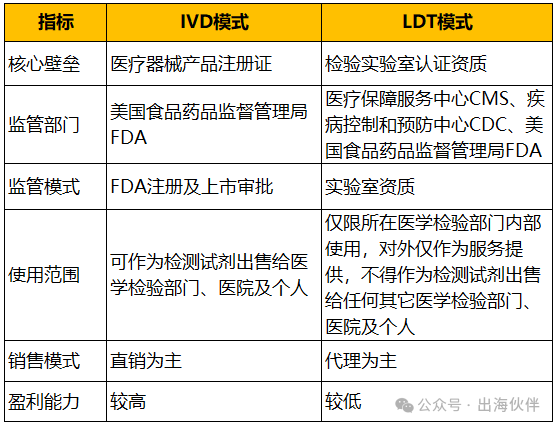
1976年,《医疗器械修正案》法案通过时,当时的LDT都比较简单,主要用于罕见疾病,对病人群体的危害很小,因此FDA豁免了对LDT的审查。高复杂性实验室可按照美国临床实验室改进修正案(CLIA)的要求,和行业标准对检测项目进行开发验证以确保检测结果的准确性,之后便可用于患者检测。但随着临床医疗和诊断科学的大力发展,过去10多年来大量不再简单的LDT涌入市场,由此带来的检测质量良莠不齐,给病人或社会带来了潜在危害,让FDA开始了将LDT纳入监管的历程。2014年,FDA发布了LDT监管指南草案《Framework for Regulatory Oversight of Laboratory Developed Tests
(LDTs)》。但后因遭到实验室和医检行业的极力反对,使之未能实施。2017年,FDA又发表了一份关于LDT政策制定的讨论文件。该文件再次表明了FDA的立场,重申将LDT作为医疗器械进行管控的决心,并表示支持以立法来解决。2020年,有国会议员便提出了VALID议案,并要求FDA对LDT进行监管。2022年,美国国会进行了迄今为止最认真的尝试之一,试图通过立法赋予对LDT的监管权。该法案被称为“验证准确的前沿体外诊断检测(VALID)法案”。该法案将为所谓的体外临床检测(IVCT)创建了一个基于风险的框架,其中包括IVD和LDT。在国会未能通过VALID法案后,FDA宣布将寻求在其现有权限下监管LDT。2023年9月29日,FDA发布了有关LDT监管的拟议规定(proposed rule)。该规定明确指出LDT属于体外诊断(IVD),而所有IVD都属于医疗器械,因此由FDA 根据《联邦食品、药品和化妆品法案》进行监管。2024 年 4 月29 日,FDA 宣布了关于LDT的最终规章,旨在明确表明这类产品将作为医疗器械受到 FDA 监管,目的是帮助确保LDT的安全性和有效性。同时,FDA还发布了一项政策,将在4年内逐步取消对LDT的一般执法自由裁量权。FDA 还针对实验室生产的某些类别的 IVD 发布了有针对性的执法自由裁量权政策。政策指出:将在 4年内对LDT的一般执法自由裁量权的逐步取消政策包括如下5个阶段:
资料来源:FDA官网https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests。根据以上LDT政策发展历程,可见,FDA对LDT监管的决心,未来对LDT的监管将渐趋严格。即把产品供应给有CLIA Lab资质的实验室,目前有些创新IVD上市的途径之一,例如Quanterix公司的其中一部分收入就来自Lab。
今年4月29日,FDA通过了监管LDT的法规,把LDT纳入了FDA监管范畴,将在未来4年逐步取消LDT的一般执法自由裁量权。FDA多年前就想管LDT,经过疫情后发现很多LDT产品数据不够准确、不够安全,可能存在造成患者伤害的隐患。通过上面的FDA监管历程,我们也能够体会到FDA的决心。根据近2年推出的法规,我们可以看到,最终FDA还是推出了LDT的监管政策。根据以上政策走向,后续FDA未来对于LDT的监管将渐趋严格。FDA上市认证主要包括510K和PMA,以510K为例,这是多数IVD企业II类产品选择的上市路径,资料齐全的情况下,审批时间4个多月。该上市途径的好处是,商用范围更广(LDT范围仅限所在医学检验部门内部使用,不得作为检测试剂出售给任何其它医学检验部门、医院及个人)。有些企业问,IVD认证流程长,LDT上线周期短,是否后续可选择LDT模式?在FDA监管趋严的背景下,对于已有LDT的企业,以IVD的逻辑去开发LDT产品,把LDT产品作为pre-IVD产品打磨,最终实现IVD产品化,可能是比较现实的路径。从大趋势来讲,LDT只能作为补充形式,不可能成为主流模式,对于拟在IVD领域深耕并开拓海外市场的企业,对于已有LDT模式的产品,做好FDA认证的准备将会是更为合理的路径。声明:本文由伙伴智能整理,欢迎关注与分享。如需转载,请注明来自 “伙伴智能”。
|
声明:
1、凡本网注明“来源:小桔灯网”的所有作品,均为本网合法拥有版权或有权使用的作品,转载需联系授权。
2、凡本网注明“来源:XXX(非小桔灯网)”的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。其版权归原作者所有,如有侵权请联系删除。
3、所有再转载者需自行获得原作者授权并注明来源。


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 
















