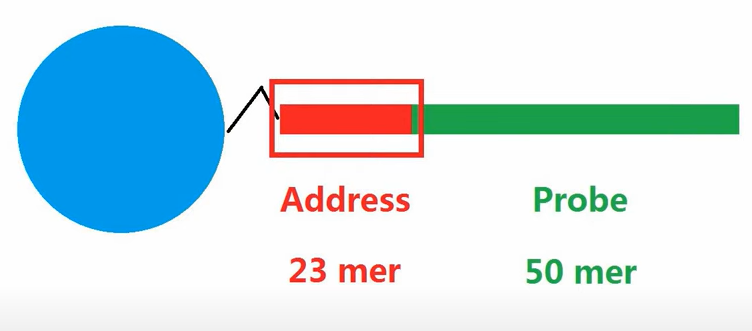
一、 illumina磁珠芯片制造原理芯片由玻璃基片和微珠组成,光蚀刻出许多微米级小孔,用于容纳微珠。每个微珠表面偶联一种序列的DNA片段(一个珠子上片段序列相同),每个微珠上有几十万个片段。
5'端address序列是标识微珠的标签序列,每种序列就是微珠的身份证号ID。 3'端probe为探针序列
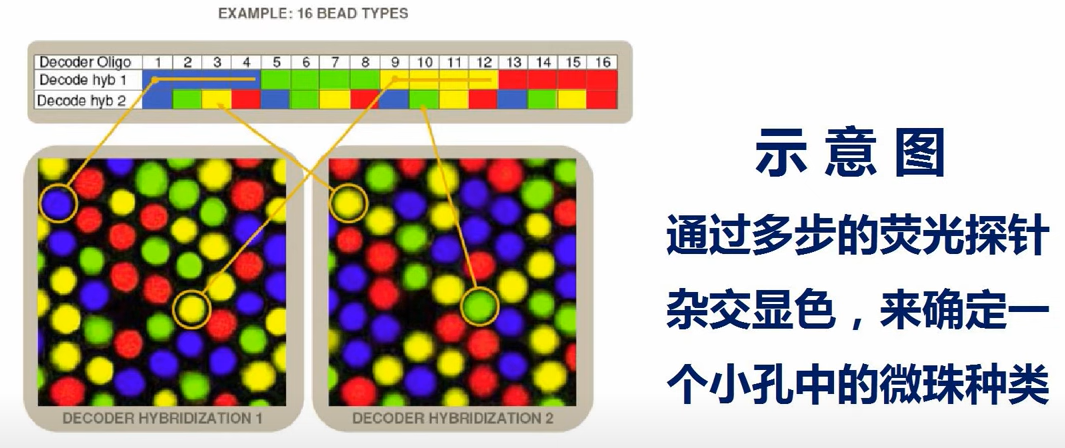
illumina芯片的制造过程:把要做芯片的几十万种微珠,按设定的比例混合好,撒到玻璃基片上,微珠随机的落入基片的小孔中。然后通过检测芯片上每个小孔中微珠的address序列,就可以知道小孔当中是哪种微珠。因为address序列和probe序列一一对应,因此就知道每个小孔中有那种probe。
因此illumina公司出厂的每张芯片都跟一个 ❝ 二、 lumi: a pipeline for processing Illumina microarray
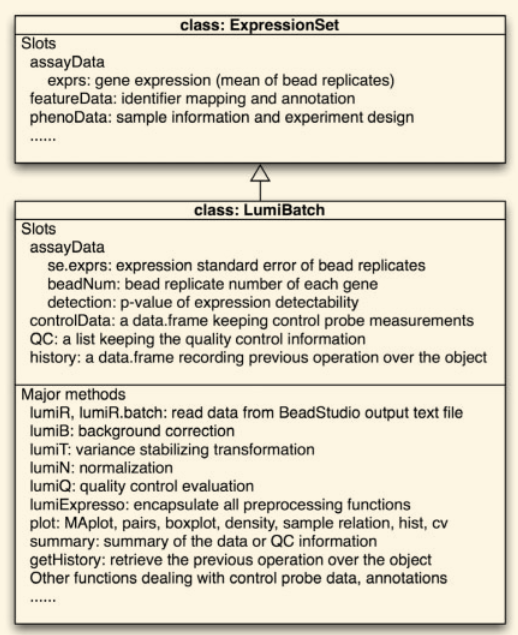
「摘要:」 illumina使用的BeadArray(磁珠芯片)技术需要不同的预处理和质控,相较于其他芯片技术。不幸的是,大多数分析软件并没有利用BeadArray 系统的独特特性,而只是结合了最初为 Affymetrix 芯片设计的预处理方法。lumi是专门为处理illumina芯片数据设计的R包,可以从Bioconductor下载获得。它包括芯片读入,质控,固定方差,标准化和基因注释部分。具体来说,lumi包 包括「方差固定变换 (variance-stabilizing transformation,VST) 算法」,该算法利用每个 Illumina 芯片上可用的技术性重复(同一序列对应约30个磁珠)。该包提供了不同的标准化方法和众多的质控图。为了更好的注释illumina数据,供应商自主的核苷酸通用标识符(nuID)用于识别illumina芯片的探针。nuID注释包和lumi处理输出的结果可以轻松地与其他Bioconductor包集成,以构建Illumina数据的统计分析的工作流程。 1 介绍illumina磁珠芯片有约30个随机定位的「重复磁珠」(具有同样的探针序列)。与其他类型的芯片相比,这种额外的设计可产生更高的置信度和更稳健的估计。然而,Illumina 微阵列设计的独特性使得预处理和质量控制步骤与其他类型的微芯片显著不同。不幸的是,到目前为止(2007),大多数分析软件都没有利用 Illumina BeadArray 系统的这种独特特性,而只是结合了最初为 Affymetrix 芯片设计的预处理方法。lumi包提供专门为illumina磁珠芯片设计的算法,并且使用现有的算法和基因注释的框架。除了支持芯片数据的现有算法外,lumi 包还包括几个独特的部分:(1) 利用 Illumina 芯片上可用的技术重复的固定方差变换 (VST);(2) 为 Illumina 微阵列数据设计的标准化算法 ;(3) 核苷酸通用标识符 (nuID) 注释包。nuID 注释包允许对每个探针进行不依赖版本和供应商的注释。nuID 还通过包括错误检查的过程对原始探针序列进行唯一且准确的编码。 2 应用2.1 预处理和质控lumi包包含一个主要类:「LumiBatch」,它继承自Bioconductor中的 「ExpressionSet类」,以实现与其他Bioconductor包的互操作性。LumiBatch类的结构如下所示,它在ExpressionSet类的基础上延伸出了三个元素:在assayData槽下的: controlData槽保存对照探针的信息,QC槽保存质控总结,history槽追踪所有在LumiBatch对象上进行的操作,可以解释数据来源。用户可以选择将BeadStudio输出的Illumina注释信息保留在LumiBach对象的featureData中。 lumi包中有几种主要的处理方式。lumiR 通过智能读取所有版本的 Illumina BeadStudio 软件的原始数据来初始化 LumiBatch 对象,并且 lumiR.batch 方法旨在读取一批数据文件。 这些方法还包括调用先前为 Affymetrix 数据设计的其他处理方法的选项。出于为更好的可视化和质控的目的,lumi 包还提供了不同类型的可视化功能。这些绘图函数可以处理表达和对照探针数据。更多详细信息请参阅教程和函数帮助文件。
2.2 注释包Illumina 注释包是使用 Bioconductor 注释工具构建的,并使用每个探针的 nuID 作为标识符。比对 TargetID 或 ProbeId 到 nuID 也包含在注释包中。由于Illumina的芯片均采用50mers,通过使用nuID通用标识符,我们可以为同一物种的不同版本芯片建立一个注释数据库。此外,nuID可以直接转换为探针序列,并用于获取最新的refSeq匹配和注释。目前所有Illumina表达芯片的注释包(包名称以“lumi”为前缀,后跟物种名称和版本号,例如lumiHumanAll.db)可以从Bioconductor下载。 3 使用案例图2 显示数据处理流程图。用于预处理的R源代码如图3所示。由于lumi包中的类是从类ExpressionSet扩展而来的,因此Bioconductor中的许多数据分析包可以直接应用于lumi产生的结果。图 2 显示了使用 lumi 包以及 limma、GOstats 和 MLInterfaces 的场景。更多的实现细节可以在lumi包的教程中找到。 总之,lumi 包提供了LumiBatch类的基础框架和相关方法来构建 Illumina 从原始数据开始到功能分析的工作流程。
三、 以GSE67936为例的原始数据处理
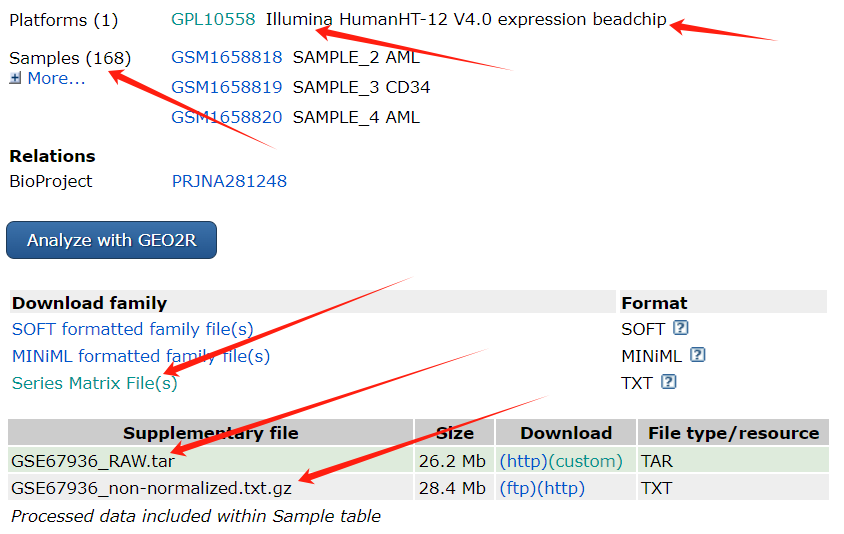
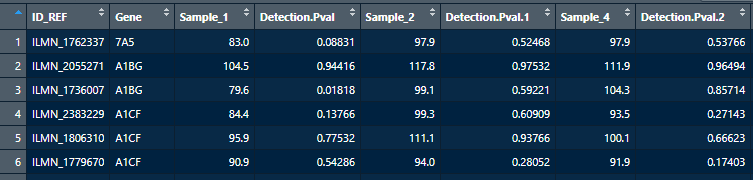
可以看到是illumina磁珠的表达量芯片,一共有168个样品。 Supplementary files 中有一个RAW原始数据的压缩包和一个non-normalized为标准化数据的压缩包。这个示例数据中的RAW.tar不可用,存储的平台的注释信息。我们读取"GSE67936_non-normalized.txt" 进行分析。 读进来的a中第一列是探针id,第二列是symbol,从第三列起每两列对应一个样本的信号值和pvalue。
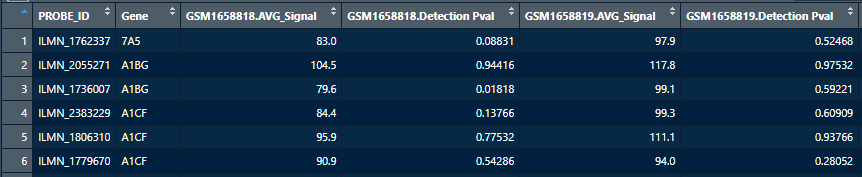
整理矩阵行名使之适用于lumiR的输入:
之后按照lumi文献提供的方法lumiR读入数据。 lumiB调整芯片背景;lumiT 对数据进行方差固定;lumiN 对方差固定后的数据进行标准化,lumiQ 评估数据质量。这些步骤可以使用封装好的lumiExpresso函数一步完成 因为这里我们处理过的lumi.N.Q已经是ExpressionSet对象了,所以可以用exprs函数提取其表达矩阵
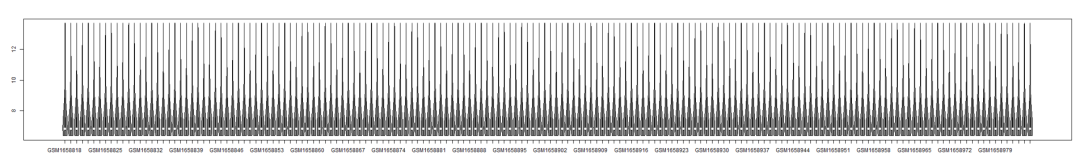
可以看到行名既不是探针id也不是symbol,还有补救方法,从lumi.N.Q@featureData@data中提取探针id,在转换成symbol 箱线图检查一下单个样本表达量分布和样本间方差齐性
之后就可以进行后续分析了 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号