10月9日,上海LDT试点文件重磅出炉
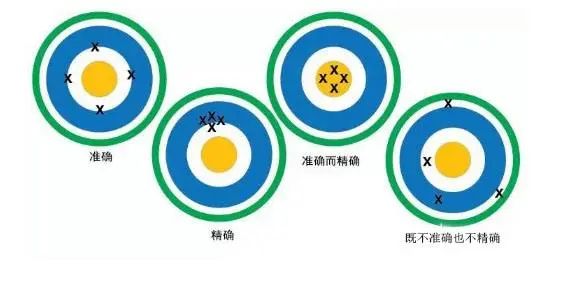
该通知将复旦大学附属中山医院、上海交通大学医学院附属瑞金医院、上海交通大学医学院附属仁济医院、上海市第一人民医院、复旦大学附属华山医院等医院列为试点综合类单位。 积极支持实验室自建检测方法(LDT)试点,有条件允许LDT项目服务于临床推广。推动各级临床医学研究中心和生物样本库建设。探索建立医学检验诊断数据质量控制体系与标准。鼓励开发“整合式”平台技术,研发高端检验检测一体化设备,攻克仪器的稳定性、可靠性、微型化和智能化等关键技术。鼓励医检业务外包,将第三方医学检验作为公立医疗机构的补充。支持检验检测产品纳入医保目录。 医学检验试剂是医疗机构用于诊断疾病,为临床提供诊疗参数的特殊物品,其检测的质量直接影响临床检验结果,从而关系到对疾病性质的定位,疾病严重程度的掌握,治疗方法的选择和预后的判断。随着LDT试点工作的逐步开展,更多个体化IVD试剂逐渐涌入市场,规模在不断扩大,为保证个体化化诊断试剂的质量,确保检测系统的有效性,需要第三方医学检验所对这些诊断体系进行检测性能的验证。 (一) 体外诊断试剂的分类 美国体外诊断产品按医疗器械风险管理,可分为Ⅰ类、Ⅱ类、Ⅲ类,Ⅰ类为豁免检测项目,美国食品药品监督管理局(FDA)批准这类试验可接受厂家性能规范,最低要求就是执行厂家的说明书,对其性能验证没有特别要求;Ⅱ类、Ⅲ类为非豁免检测项目,与Ⅰ类相比,Ⅱ类、Ⅲ类属中度和高度复杂的检验项目,目前实验室进行的绝大多数个体化检测均属于非豁免检测项目,应进行规范的性能验证。在我国,根据《体外诊断试剂注册管理办法》,跟美国大体一致,体外诊断试剂也可分为Ⅰ类、Ⅱ类、Ⅲ类,其中Ⅱ类、Ⅲ类要求进行性能验证,国内现有的个体化诊断项目也绝大多数属于此类。 (二) 性能验证内容 检测体系性能验证的内容有多种,包括准确度、精密度、线性、可报告范围、检出限、参考区间、灵敏度、特异性等。 1. 准确度评价 准确度是指测试结果与参照值间的一致程度,它不能直接以数值表示,通常以不正确度来间接衡量,我们可以将正确度评价理解为是对检测体系系统误差的评价,也可理解为是偏倚试验。 准确度评价的方法主要有两种,一是与标准物质进行比较,使用待评估项目对已知标准值的标准物质进行分析,将检测结果与已知标准值进行比较。第二种方法是同时使用待评估项目与标准方法或参考方法对同一批次样品进行分析,然后将不同方法得到的结果进行对比分析。 准确度在定性测定,通常是将其待验证方法与一公认或实验室已在使用的方法同时检测日常工作标本,然后比较两种方法之间结果的差异,不一致的结果再用第三种或金标准方法确认。
2. 精密度评价 精密度是指重复检测条件下,获得的独立测量结果间的一致程度,它和准确度一样也不能直接以数值形式衡量,往往以不精密度表示。我们可以将精密度评价理解为是对检测体系随见误差的一种度量,常以标准差的形式表示,标准差越小精密度越好。 若想验证新检测体系的精密度可通过对一定样本进行重复测量分析而直接得到结果。一般来说有两种方法,如这种待评估方法为标准方法,则使用该方法直接重复检测标准物质,直接比较检测结果的标准差和标准方法中规定的重复性限;如已有其他标准方法,则将待评估方法直接与标准方法进行比较,两种方法同时对一系列的标准物质或临床常规样品分别进行分析,对两种方法检测结果的标准差或方差是否有统计学差异。 精密度在定性可理解为,一份阳性或阴性标本,多次重复检测,其出现阳性或阴性结果的次数。当然,阳性样本不宜完全采用强阳性,而应采用接近检测临界水平的样本。
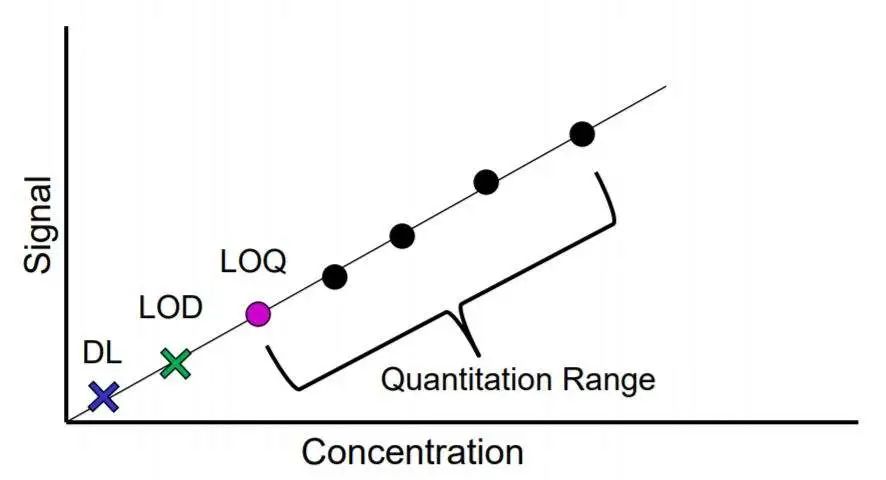
3. 线性与可报告范围验证 线性是指在检测体系的检测范围以内,测量值与预测值之间的关系。需要说明的是线性与准确度密不可分,高准确度是线性分析的必要条件。 线性分析有两种常见方法,我们可以直接分析已知浓度样品也可将一定浓度样本进行系列稀释,根据稀释因子来研究检测值与逐步下降的预期估计浓度之间是否存在线性。 可报告范围是能够报告的可靠的最低和最高检测结果,一般由厂家/ 方法建立者提供,验证实验室可通过“多点法”进行简单验证,CLSI推荐至少使用4个,最好是5个不同浓度水平样品进行验证。这些不同浓度样品中需包含一个接近检出限的零水平浓度和一个接近或稍高于最高检出限的浓度,重复测量4次,以测量值均值为y轴,指定值为x轴绘制图形进行分析,观察各点是否通过直线。
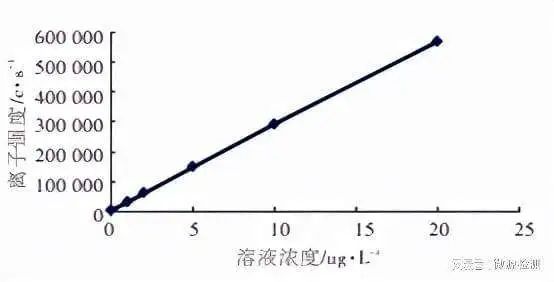
4. 检出限(LoD)验证 检出限是指可被检测体系检出的最低检测浓度,又称最小检测浓度或检测底线。建立、验证LoD时,需同时建立、验证空白检测限(LoB,一定概率下测量空白样本时可能得到的最高检测结果)。 检出限的建立一般由厂家、方法建立者完成,实验室如果只是想验证分析结果是否与厂家/ 方法建立者给出的检出限是否相符,可以根据一个简单的程序完成:如已有厂家/ 方法建立者提供的空白检出限,则首先需对LoB进行验证,一般的做法是对一份空白样本重复检测20次,若超过LoB的个数不超过3则该LoB可用。之后重复测定浓度为LoD的样本至少20次,计算大于LoB的测量结果所占比例是否符合要求(β=5%),符合要求则验证通过,不符合要求则联系厂家/ 方法建立者,或自行建立LoD。若厂家/ 方法建立者未提供LoB,则需自行建立LoB后再进行下一步的LoD验证。 需要特别指出的是,很多人常将检出限和分析灵敏度混淆,分析灵敏度是指检测方法分析出待测样本小变异的能力,通常用校准曲线的斜率表示(横坐标为浓度,纵坐标为测量信号),斜率越大,直线越陡,灵敏度也高。
5. 参考区间验证 参考区间是指上、下参考限之间的参考值分布范围,医学参考值区间通俗地讲就是“正常人”各项生理指标正常波动的范围,主要用于划分正常与异常人群。建立时需抽取足够数量的“正常人”作为研究对象,进行统一而准备的测定。所需要的数量一般要求大于100例;“正常人”不是指任何一点小疾病都不能有,而是指需排除影响研究指标的疾病因素;测定时方法、仪器、试剂、操作熟练程度等都要统一。 参考范围的建立一般由厂家/ 方法建立者完成,实验室如果只是想验证厂家/ 方法建立者给出的检出限是否适用,可以通过三个方法进行验证:第一,直接分析法:直接比较厂家/ 方法建立者进行参考范围建立研究时的相关分析、分析 前因素是否与本实验室一致。如参考人群的地理因素、人口因素等是否与本实验测试对象人群一致,如大体一致,可认为相符并直接使用该参考范围,但这种方法比较粗糙;第二,验证实验室可检测少量本实验室测试对象(如n=20),将这部分参考个体检测值与厂家/ 方法建立者的大型初始研究进行比较。如果这个20个参考个体中检测值落在厂家/ 方法建立者提供的参考范围以外的个数≤2(测试结果的10%),则本实验室可使用该参考范围;如20个参考个体中检测值落在厂家/ 方法建立者提供的参考范围以外的个数≥3,验证实验室需重新收集参考个体进行检测,若超过厂家/ 方法建立者提供的参考范围以外的个数≤2,那么本实验室可使用该参考范围,反之仍≥3则需重新分析两组人群的生物特征差异,考虑自行建立参考范围。第三,验证实验室检测更大量本实验室测试对象(如n=80),将这部分参考个体检测值与厂家/ 方法建立者的大型初始研究进行比较。这个方法因参考个体数量的增加而更具科学性。
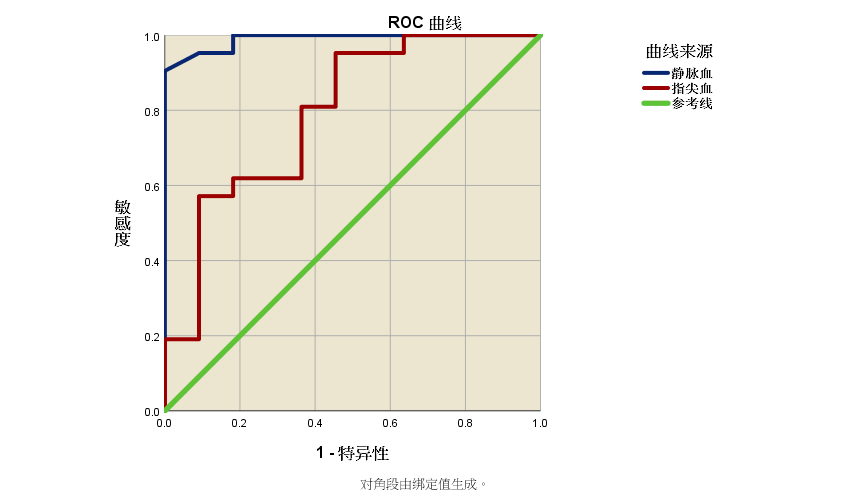
6. 灵敏度、特异度验证 灵敏度和特异度是反映诊断试验准确性的两个最基本的统计指标。灵敏度表示实际患病者按检测结果正确判为有病的概率,特异度表示实际未患病者按检测结果正确判为没病的概率。通常情况下,几乎没有一种检测的特异性和敏感性能够达到100%或1.0,两者同时提高比较困难,提高灵敏度往往要同时降低特异度,反之亦然,如何根据临床测量选择诊断标准应根据实际情况而定,对于筛查试验一般是希望灵敏度高一些,而对于确诊试验则希望特异度要高一些,诊断灵敏度和特异度值一般由厂家/ 方法建立者提供,对于一个临床分子诊断实验来说,可通过绘制ROC(receiver operating characteristic)曲线,确定诊断阈值(cut-off),得到合适的敏感度和特异度。ROC曲线又称受试者工作特征曲线,在临床检验领域,被用于诊断性能验证中不同检验方法的比较和检测项目临床准确性的评价,以及检测临界值的确定。它描述的是敏感性(真阳性率,TPR)和假阳性率(1-特异性,FPR)之间的关系,并以FPR为横坐标,以TPR为纵坐标绘制曲线。具有优秀的诊断性能的检测敏感性和特异性都很良好,ROC曲线越向着左上角靠近,曲线下面积(AUC)就越大,其临床检验的准确性越好。诊断阈值的选取可根据实际情况权衡后在ROC曲线上取点,如诊断灵敏度和特异度都同等重要则可选取斜率为45°切点位置附近的点为诊断阈值。 诊断灵敏度、特异度验证需要收集一定数量的经确诊的患者样本及阴性样本,根据厂家/ 方法建立者确定的诊断阈值,进行过阴/ 阳性判定,计算诊断灵敏度与特异度,并与厂家/ 方法建立者提供的诊断灵敏度和特异度对比,观察是否有统计学差异。
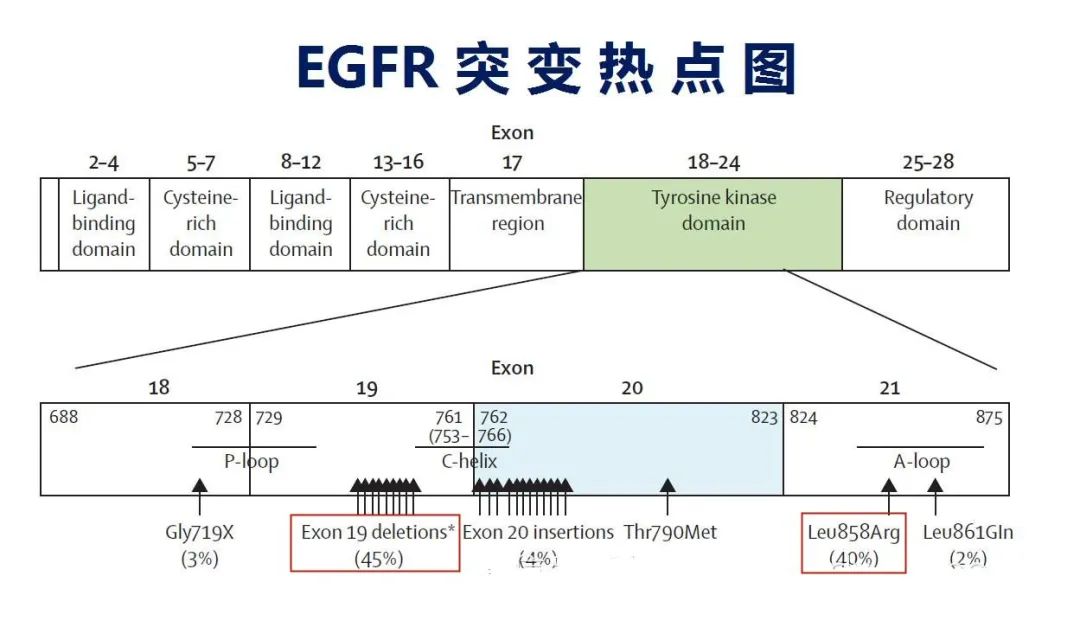
(三) 个体化检测体系性能验证的特殊性 体外个体化诊断方法多为中/ 高度复杂的非豁免方法,其检测性能验证内容和其他体外诊断方法大体一致,但是也有其特殊性。 目前个体化检测体系性能验证的主要指标:根据国家食品药品检定研究院相关资料,个体化突变检测体系性能验证的主要指标包括:重复性/ 精密度、准确度(突变型的检测能力)、分析特异性(如正常野生型基因组与突变型之间的区分能力)、检出限(每个反应能检出的最小突变拷贝数)、选择性(突变型与野生型不同比例设定)。 关于标准物质:个体化突变检测体系进行准确度、精密度等性能验证时所使用的参考物质为各种突变型和野生型的质粒或突变细胞株,如EGFR突变检测国家参考品包括了29种EGFR突变体参考品、5种野生型参考品、1份重复性EGFR突变体参考品;EGFR突变细胞株SW48、H1975、H1650包含了突变位点19号外显子缺失、18号外显子G719S和20号外显子T790M、21号外显子L858R四个突变位点。
目前个体化检测体系性能验证存在的主要问题: ①分析特异性欠佳,部分试剂盒在检测不同突变型时存在交叉反应,正常提取浓度的野生型基因组会对部分试剂盒产生明显的非特异性反应; ②诊断阈值(参考范围)的确定较难,因为目前对肿瘤靶向药物使用阈值的确定研究尚未明确; ③突变检测性能验证存在难点,如评价临床突变诊断敏感性和特异性需要足够多的受试者包括野生型个体和突变型个体,因此患病率低、外显率低、遗传异质性或其他罕见特征的疾病对应的研究设计相对较难。 因此需制定合适的受试者选择方案(纳入/ 排除标准),避免引入偏倚。当评价一种具有低发病率或者从检测到发病有一段较长时间的疾病时,可能不便于评价目标人群的诊断性能,此时可使用症状明显的受试者大概估计检测的敏感性,使用疾病易感者或超过疾病发病年龄尚未受到影响的人来评价检测的特异性,但结果解释应该估计偏倚。产前诊断检测体系性能验证的最好方式是前瞻性研究,以产前诊断-产后随访的形式进行。而另一些检测,如HIV-1耐药突变检测其诊断效能验证也比较棘手,主要原因是没有金标准来确证该突变是否确实存在。也许有的同道会推荐用测序的方法,但是在实际情况下,病毒往往不是单一的突变型或野生型存在,对于那些混杂型突变(突变型和野生型共存),尤其是突变型病毒仅占病毒总拷贝数20%以下的时候,测序就会漏检,因此目前还没有很好的方法解决这类检测诊断效能验证的问题。 |


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号