短串联重复序列(STR)是一种短DNA序列基序,通常为2到6个碱基对(bp),在基因组中的给定位置连续重复。STR约占人类基因组序列的7%,具有高度多态性,通常在无关个体之间长度不同。异常长或“扩展”的STR等位基因是人类群体中一类重要的致病性变体。迄今为止,已有40多个基因的STR扩展被证明会导至遗传性疾病,其中大多数表现为原发性神经或神经肌肉表现。这些疾病包括亨廷顿病(HD;HTT)、脆性X综合征(FXS;FMR1)、遗传性小脑共济失调(RFC1、FXN和其他)、强直性肌营养不良(DMPK和CNBP)、肌阵挛性癫痫(CSTB、SAMD12、STARD7和其他),以及C9orf72相关的额颞叶痴呆和肌萎缩侧索硬化(ALS)。据估计,这>40个STR相关的神经遗传疾病中,每10万人中约有1至10人感染其中一种,它们的总体患病率很高。此外,与STR扩展相关的疾病列表仍在增加,许多致病性STR基因最近已被描述。 鉴于(i)STR扩展障碍的广泛多样性和总体高患病率,(ii)涉及的基因数量众多,(iii)新基因的频繁识别,(iv)致病性STR扩展等位基因的大小和序列构象多样性,以及(v)我们对其基本生物学的理解存在许多差距,人们越来越需要改进的STR分子表征方法。已建立的分子技术,例如,Southern杂交和重复引物聚合酶链反应(RP-PCR),相对缓慢、劳动密集且不精确,需要对每种不同的STR分别使用特定的引物/探针进行检测。当多个不同的STR扩展可以表现为相似的表型(基因座异质性)时,这成为一个问题,并且是对新识别的STR基因进行检测的主要障碍。下一代测序(NGS)对STR扩展的分析有一定的实用价值。然而,许多致病性STR扩展的大尺寸、低序列复杂度和高GC含量使得它们难以通过短读NGS平台(例如Illumina)进行分析。 Oxford Nanopore Technologies(ONT)和太平洋生物科学公司(Pacific Biosciences)的长读测序技术可用于对大型复杂STR扩展进行基因分型。同时分析重复位点的DNA甲基化是这些技术的另一个优势。然而,在这两种平台上进行全基因组分析的成本仍然高得令人望而却步。最近开发了一种基于cas9的STR基因座定向富集和长读测序方法。然而,这与现有的分子技术有着相同的局限性,因为每个不同的STR都需要一组独特的cas9导向RNA,需要仔细设计和实验优化。定向长读测序的另一种方法是ONT的“ReadUntil”功能,通过该功能,可以对ONT测序设备进行编程,以便在测序实验期间识别和接受/拒绝特定的DNA序列片段。目标选择是完全灵活的,除了标准的库准备外,不需要额外的实验室过程。 近日,来自澳大利亚和以色列的研究团队在SCIENCE ADVANCES杂志上发表了一篇题为“Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing”的文章。在文章中,研究团队使用一个由37个与神经和神经肌肉疾病相关的STR基因座组成的定制panel,使用可编程的靶向长读测序和Oxford Nanopore的ReadUntil功能,在一次分析中对所有已知的神经源性STR进行平行基因分型。这种方法可以正确诊断一个小队列(n=37)中的所有个体,包括患有各种神经遗传疾病(n=25)的患者。有针对性的长读测序解决了大而复杂的STR扩展,这些扩展会混淆已建立的分子检测和短读测序,并识别非标准STR基序构象和内部序列中断。研究团队还观察到已知和未知致病基因的STR等位基因多样性,这表明长阅读测序将重新定义重复性疾病的遗传格局。研究证实了可编程ONT测序在STR扩展障碍基因诊断中的分析有效性,并展示了这种方法的诸多优势。
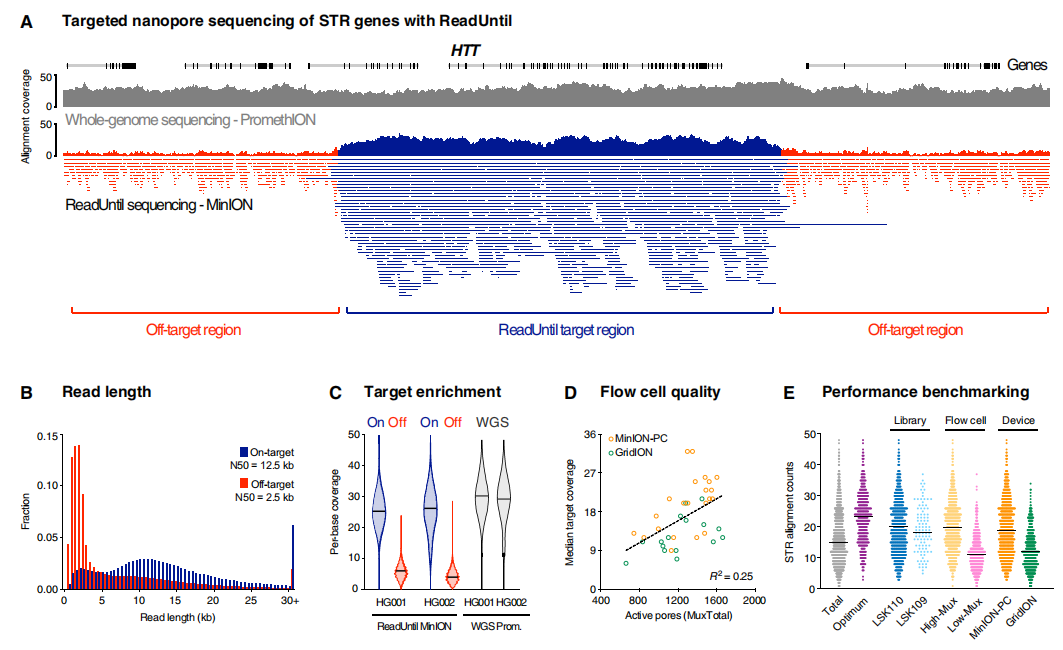
图片来源:Science Advances 主要内容 致病性STR的可编程靶向nanopore测序 ONT的ReadUntil功能有可能对所有已知致病性STR基因座进行简单、经济高效的测序,但这是一项基本上未经测试的技术。为了评估ReadUntil在STR分析中的应用,研究团队设计了一个定制panel,包含所有已知的与原发性神经和神经肌肉疾病相关的致病性STR扩展基因。对于每个基因,整个位点,包括两个方向的50kb侧翼序列都被包含。该panel包括一系列额外的临床信息位点,总共覆盖了约50.5 Mb,相当于人类参考基因组的约1.6%。 研究团队使用开源软件包Readfish对37个基因组DNA样本进行了靶向测序。结果显示,与目标读取(N50=12.5 kb)相比,非目标读取(N50=2.5 kb)的读取长度持续减少,这表明成功地拒绝了目标区域之外的片段。这导至目标区域内测序深度的中位数增加了4.6倍,在整个队列中产生了约9到40倍的目标覆盖率。
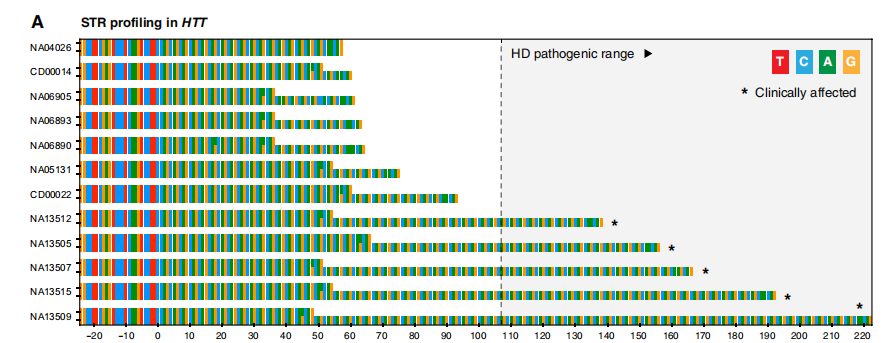
使用ONT ReadUntil对致病性STR位点进行定向测序。 图片来源:Science Advances 可编程靶向STR测序的有效性和实用性 为了确定靶向纳米孔测序分析的有效性和实用性,研究团队分析了一系列患者来源的参考DNA样本和神经遗传疾病患者(n=25)的DNA样本,包括HD(n=5);FXS(n=2);小脑共济失调、神经病变和前庭无反射综合征(CANVAS;n=6);Kennedy脊髓和延髓肌萎缩(SBMA;n=1);强直性肌营养不良1例(DM1;n=5);神经元核内包涵体病(NIID;n=1);弗里德赖希共济失调(FRDA;n=2);ALS(n=1);脊髓小脑共济失调1型(SCA1;n=1);眼咽肌营养不良症(OPMD;n=1),以及已知的突变前携带者(n=6)和未受影响的个体(n=6)。样本已在经认证的遗传病理学实验室或使用标准方法进行独立分子检测。 1. 亨廷顿病 HD是一种常染色体显性遗传神经退行性疾病,由HTT基因中≥36个“CAG”基序STR扩展引起,在≥40拷贝时完全外显。STR扩展大小与疾病严重程度相关,扩展等位基因内缺乏特征性的“CAA”中断基序也是如此。因此,HD的基因诊断需要准确的等位基因特异性STR测定和内部序列测定。 ReadUntil靶向测序在HTT外显子1的STR位点产生了足够的数据,足以对每位患者两个STR等位基因进行分期和组装,在整个队列中识别12到74个拷贝的CAG重复序列。在所有受HD影响的患者(n=5)中,检测到一个长度在已知致病范围内的扩展STR等位基因,而在未受影响的个体(n=32)中未检测到致病性扩展。通过ONT测序确定的扩展和非扩展STR等位基因的长度与临床试验密切一致。在所有HD患者和非HD患者的两个STR等位基因中均检测到单一CAA中断,后者中的六个个体存在双重CAA中断。HD样综合征也可能由靶向测序panel中包括的其他STR基因引起(即C9orf72、PRNP、JPH3、TBP、ATXN8、FXN和ATN1)。这些基因的平行测序显示,所有HD患者的STR等位基因均在健康范围内。在不需要额外的分子检测的情况下,排除这些基因中共同发生的STR扩展的能力,是多基因分析的一个明显优势。
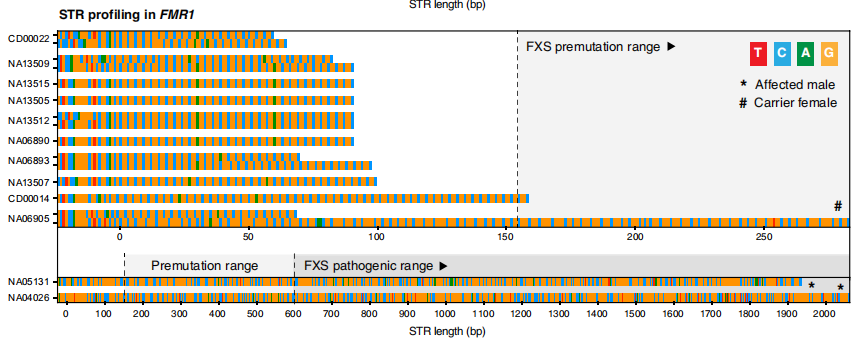
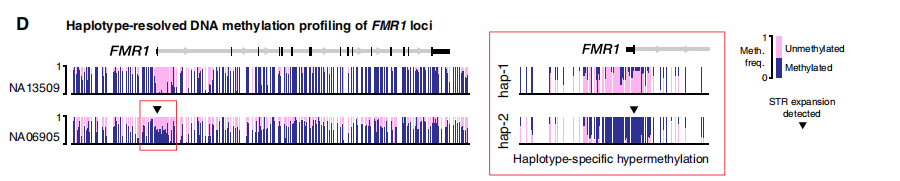
HTT的单倍型解析组装。图片来源:Science Advances 2. 脆性X综合征 FXS是遗传性智力残疾的最常见原因,也是男性自闭症谱系障碍的单基因原因。FXS是由X染色体连锁基因FMR1内的大(>200)“CGG”STR扩展引起的。DNA甲基化(5mC)也与FMR1相关疾病的致病机制有关,扩展的等位基因通常表现为启动子高甲基化和FMR1沉默。因此,除了STR大小和内部序列测定外,FMR1相关疾病的完整基因诊断还需要DNA甲基化分析。 在FMR1 5′非翻译区(UTR;外显子1)的STR位点,在整个队列中成功组装成20到654个拷贝的CGG STR。扩展和非扩展等位基因的长度与临床试验密切一致。ONT测序的结果正确地将受影响的男性个体(n=2)和女性携带者(n=1)与未受影响的个体区分开来,并将突变前等位基因(n=2)与完全致病性STR扩展区分开来。DNA甲基化分析显示,两个FXS男性均存在FMR1启动子区域的高甲基化,都有完整的STR扩展(CGG654和CGG606),局部CpG位点的甲基化频率中位数>75%。相比之下,在具有正常和突变前STR等位基因的男性中,启动子CpG甲基化频率较低(中位频率<25%)。除了STR大小和中断状态外,在一个简单的单一分析中获得单倍型解析的DNA甲基化图谱的能力是此方法的一个明显优势。
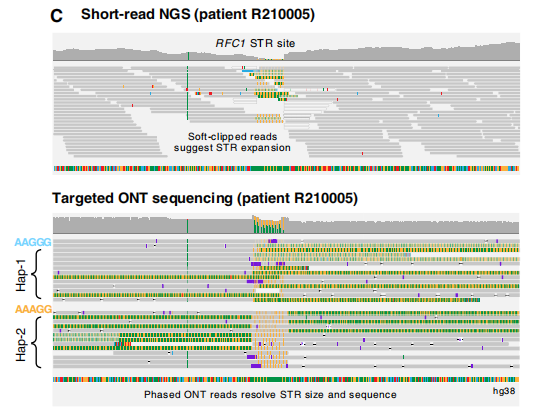
FMR1的单倍型解析组装和DNA甲基化分析 图片来源:Science Advances 3. 小脑共济失调、神经病变和前庭反射综合征 CANVAS是一种神经退行性运动障碍,在大多数情况下,由RFC1基因中的“AAGGG”STR扩展引起。除了典型的致病基序“AAGGGexp”,罕见的“ACAGGexp”基序和混合的“AAAGG10–25AAGGGexp”都被认为是致病的,而其他各种观察到的构象的致病性目前尚不清楚。 ReadUntil靶向测序单倍型解析STR组装显示,在RFC1第二内含子内的STR位点中有各种不同的五核苷酸重复,大小从8到1070个拷贝不等,与Southern杂交和/或RP-PCR的分子检测有很强的一致性。在6名CANVAS患者中的5名患者中检测到致病基序(AAGGG410–1070)的大型双等位基因STR扩展,但在未受影响的个体中未检测到(n=21)。在CANVAS的一名剩余患者中,在RFC1 STR位点检测到一个大的(~5-kb)双等位基因扩展。ReadUntil靶向测序很容易区分这两个STR等位基因,识别出不同的“AAGGG1010”和“AAAGG960”。这突出了长读序列在分析大型复杂STR方面的实用性。
致病性STR位点RFC1的单倍型解析组装。 图片来源:Science Advances 4. SBMA、DM1、NIID、FRDA、ALS、SCA1和OPMD 为了进一步证明靶向ONT测序的广泛实用性,研究团队分析了SBMA(n=2)、DM1(n=5)、NIID(n=1)、FRDA(n=2)、ALS(n=1)、SCA1(n=1)和OPMD(n=1)患者。在所有病例中,此技术都能正确地对相关STR进行基因分型。。 综上所述,上述结果证明了使用靶向ONT测序对神经源性STR基因座进行精确、单倍型解析的大小测定、序列测定和DNA甲基化分析。这为STR扩展障碍的遗传诊断建立了分析有效性,并突出了该方法的诸多优势。 解析STR多样性 STR序列具有高度多态性,但由于目前基因分型方法的局限性,其真正的多样性可能被低估。临床解释依赖于区分致病性等位基因和健康个体中遇到的STR多样性的能力。通过确定每个受试个体中每个疾病相关STR位点的每个等位基因的大小和序列,靶向测序分析提供了有价值的数据,有助于确定人类群体中STR的遗传格局。 研究团队在队列(n=37)中观察到了一系列不同的STR等位基因。临床上未受影响个体的STR大小差异在五核苷酸重复序列基因RFC1(8到324个拷贝)、DAB1(8到541个拷贝)、BEAN1(10到119个拷贝)、SAMD12(13到113个拷贝)和STARD7(10到102个拷贝)中最为明显。 虽然此研究没有包含足够大且无偏的队列来得出一般结论,但很明显,有针对性的长读测序将有助于描述目前未被充分认识的STR等位基因多样性,并可能重新定义几个基因的非致病性等位基因的特征。此外,它将有助于更详细地研究致病性重复扩展等位基因的基因型-表型相关性和潜在疾病修饰因子,例如中断。
RFC1中序列多样性 图片来源:Science Advances 总结和讨论 研究团队使用可编程的靶向长读测序和Oxford Nanopore的ReadUntil功能,在一次分析中对所有已知的神经源性STR进行平行基因分型。我们的方法能够从一系列预先确定的候选基因中,对STR位点进行精确、单体型解析的组装和DNA甲基化分析。这种方法可以正确诊断一个小队列(n=37)中的所有个体,包括患有各种神经遗传疾病(n=25)的患者。有针对性的长读测序解决了大而复杂的STR扩展,这些扩展会混淆已建立的分子检测和短读测序,并识别非标准STR基序构象和内部序列中断。这项研究证明了可编程靶向nanopore测序在STR扩展障碍基因诊断中的有效性和实用性。与现有的单基因分子技术不同,此方法能够在单一靶向分析中对所有已知的神经源性STR位点进行无偏和序列测定。 ReadUntil在手持小型设备上实现了与台式设备上的全基因组nanopore测序类似的目标覆盖率,除了降低资本成本、数据存储和计算要求外,每个样本的价格降低了3倍以上。鉴于这些能力,研究团队建议使用ONT ReadUntil进行靶向测序,以满足对STR扩展分子特征的改进方法的迫切需求。 虽然靶向测序不适合在没有事先证据的情况下发现基因,但ReadUntil测序的灵活性非常适合分析数十个/数百个候选基因/区域,例如通过受影响家族中的连锁图谱确定的基因/区域。此外,重复扩展不一定是这种方法发现的唯一致病性变体,有针对性的长读测序也适用于检测其他类型的结构变异。研究团队预计,这将是一种发现STR基因的有力方法,并为未来许多以前未解决的病例提供分子诊断。 |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号